

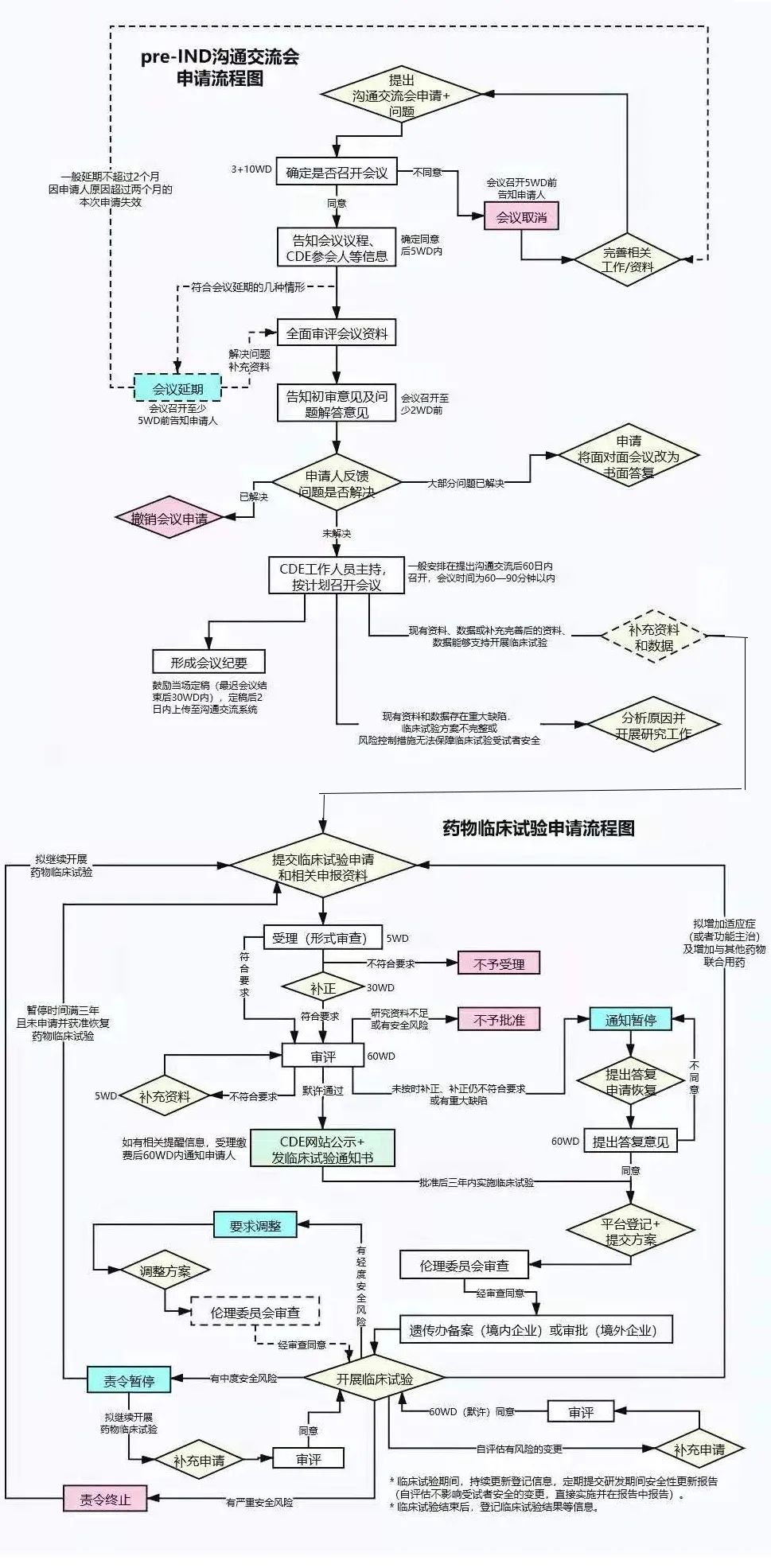

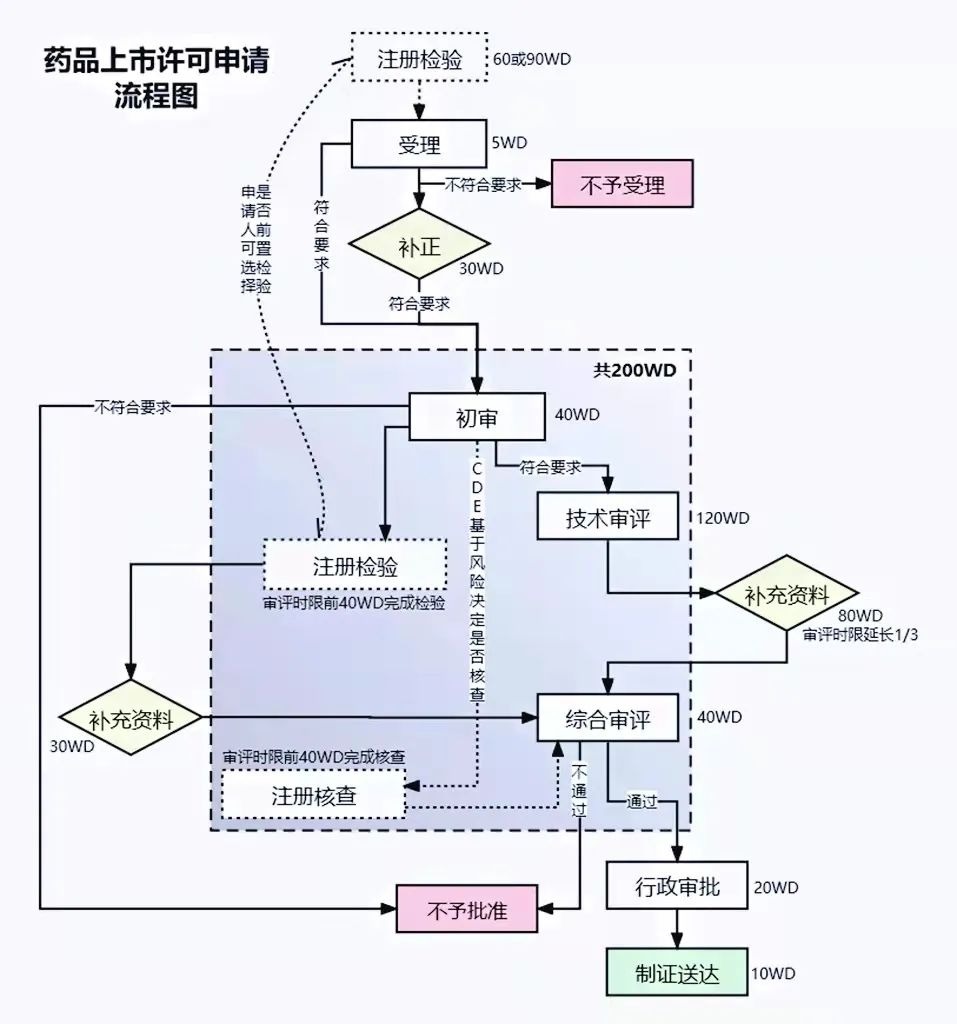
国内药品注册沟通交流制度介绍与思考
一、概述
二、沟通交流制度的演变
三、目前沟通交流分类及要求
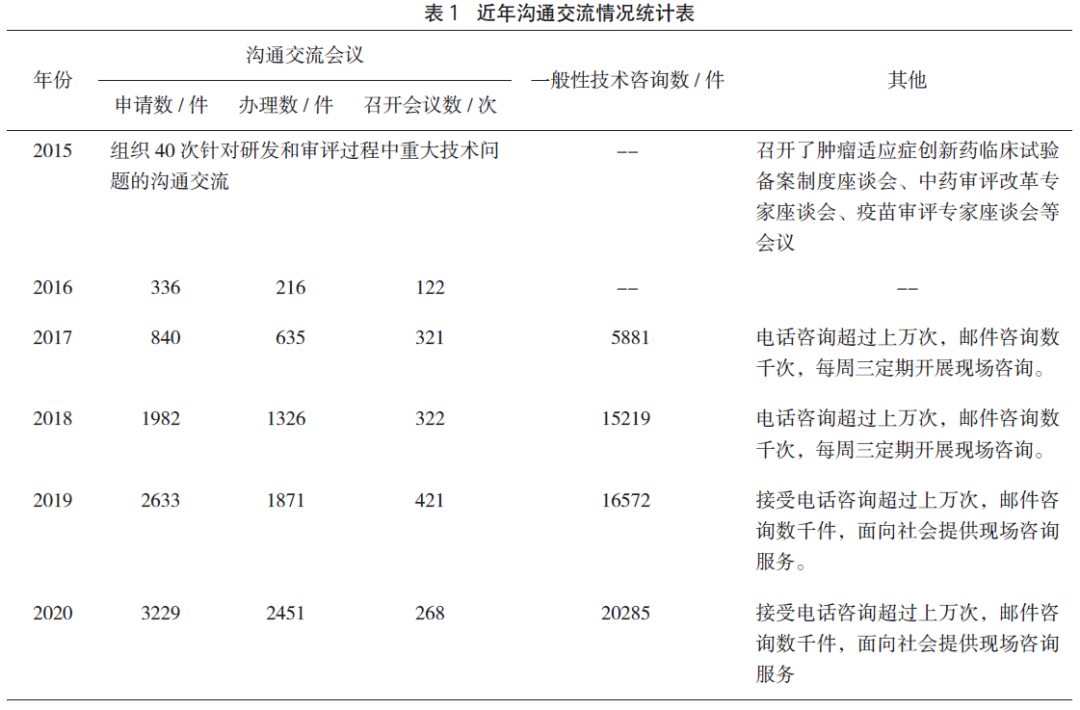
四、沟通交流情况
五、存在的常见问题
六、技术要求
七、经验与启示








编辑:小果果,转载请注明出处:https://www.cells88.com/zixun/zcfg/35229.html
免责声明:本站所转载文章来源于其他平台,主要目的在于分享行业相关知识,传递当前最新资讯。图片、文章版权均属于原作者所有,如有侵权,请及时告知,我们会在24小时内删除相关信息。
说明:本站所发布的案例均摘录于文献,仅用于科普干细胞与再生医学相关知识,不作为医疗建议。
 微信扫一扫
微信扫一扫  支付宝扫一扫
支付宝扫一扫 