

✦
在过去的几年里,细胞和基因疗法的临床管道一直在快速增长,而 FDA 现在已经准备好了,这要归功于一个新的、专门的超级办公室的建立。


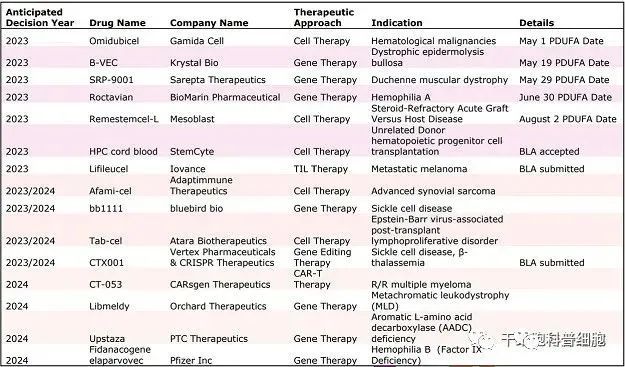
美国再生医学联盟 (ARM) 公共事务主管斯蒂芬·梅杰斯 (Stephen Majors) 6月12日对媒体表示:“今年可能总共有大约 10 项关于细胞和基因疗法的监管决定,其中仅针对罕见遗传病的基因疗法就有多达 5 项决定。” 这些可能包括几个潜在的第一,包括第一个 CRISPR 疗法——Vertex Pharmaceuticals 和 CRISPR Therapeutics用于治疗镰状细胞病的exa-cel ;第一个治疗杜氏肌营养不良症 (DMD) 的基因疗法——Sarepta Therapeutics 的 SRP-9001,它可能成为第二个通过该机构加速审批途径获得批准的基因疗法。 McKinsey & Company 再生医学公司的合伙人之一 Simon Alfano 表示,他注意到在过去十年中,临床开发中的细胞和基因疗法数量显着增加。 “目前美国有 1,000 多种细胞和基因疗法处于临床开发阶段,其中 3,000 多种处于临床前开发阶段,”Alfano说道。 “根据过去的临床成功率,”他继续说道,“这种水平的临床活动表明新细胞和基因疗法的推出将在本世纪下半叶加速。” Majors 说,现在 FDA 面临跟上步伐的压力。
启动超级办公室
FDA细胞和基因治疗产品办公室(OTP)于2023年3月正式成立,是FDA生物制品评价和研究中心(CBER)的第一个超级办公室,下设6个分办公室、14个部门和33个分支机构。FDA 超级办公室共有三个,旨在简化细胞疗法审批的工作流程,以提高机构内部的效率,造福行业和患者。 “我们目前正在填补 OTP 内的关键职位,其中包括广泛的相关科学领域和医学专业,包括大量将属于高级学位类别的雇员,”OTP 代理主任 Celia Witten 告诉BioSpace。总的来说,超级办公室预计将有 500 多名员工。 CBER 主任 Peter Marks 为 FDA 准备好迎接即将到来的细胞和基因治疗提交热潮进行了广泛的宣传。 “FDA 准备评估与拟议的临床研究或使用研究性细胞或基因治疗相关的任何 IND 申请,并根据预期的潜在未经证实的益处确定拟议的研究或治疗的风险是否合理,”Peter Marks谈道。 然而,包括 Marks 在内的该机构意识到,当前的细胞和基因疗法监管框架需要更新,以更好地应对这些产品带来的独特挑战。 该机构“致力于开发一种监管范式,以促进安全有效的基因疗法的开发,特别是对于需求未得到满足的罕见疾病,”Witten 说。 据CBER的专家称,FDA 更好地支持细胞和基因疗法的意图是推进处方药用户费用法案( PDUFA )最新重新授权的一部分。具体而言,作为PDUFA 讨论的一部分,FDA承诺为 CBER 现有的细胞和基因治疗项目增加资源。 根据 2020 年 9 月 FDA 行业会议的总结,这些细胞产品开发的快速增长导致“应用数量持续不断增加,满足了对各种不同和复杂产品的要求”。“这种增加的工作量迫使 CBER 限制赞助商互动,将许多会议转换为‘仅书面答复’,并限制外部互动,以及其他影响。” Majors 补充说,该组织从其成员那里听说与 CBER 的互动“时不时地”出现延迟。 代表 Brett Guthrie (R-KY) 和 Anna G. Eshoo (D-CA) 于 2023 年 3 月致函该机构,提出了对临床持有增加的担忧,即 FDA“最近发布了推迟拟议的临床调查或暂停细胞和基因疗法的持续调查”。 在最近的美国基因与细胞治疗学会 (ASGCT) 会议上,Marks 表示,根据 PDUFA VII 的授权,CBER 可以聘请多达 125 名审稿人,其中大部分“将进入细胞和基因治疗领域”。 Premier Research 细胞和基因治疗副总裁 Kenneth Ndugga-Kabuye 表示,根据 PDUFA VII 规定的招聘承诺和其他职责促使该机构将 CBER 的前组织和高级治疗办公室 (OTAT) 转变为新的 OTP。“创建办公室是为了提高响应能力、适应能力和灵活性,”他说。
使监管范式现代化
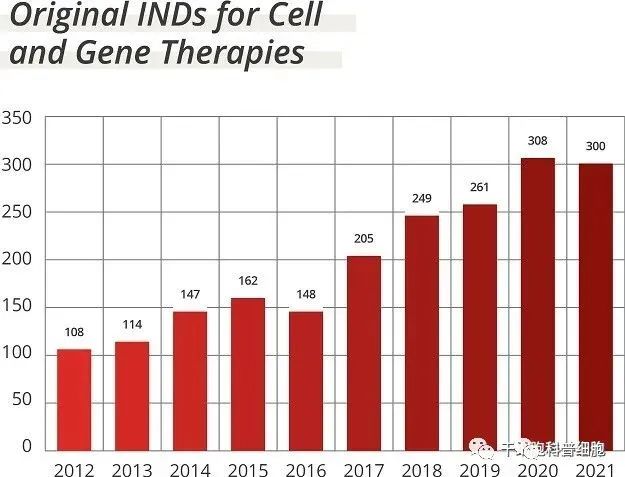
然而,OTP 的建立只是满足对细胞和基因疗法监管审查日益增长的需求的第一步。 据 Alfano 介绍,自 2020 年以来,早期管道资产的数量增长了 50% 以上,而目前正在进行 100 多项后期 III 期试验和 600 项 II 期试验。 其中一些疗法已经进入市场,自 2010 年首次批准以来,CBER 共批准了29 种细胞和基因疗法。而且这一步伐不太可能很快放缓。 自 2017 年以来,细胞和基因疗法的研究性新药 (IND) 申请数量增加了一倍多(见图),FDA 在其要求预算增加 10% 的理由中表示,这是乔·拜登总统 2024 财年预算的一部分提议。
Premier Research 业务部门 Premier Consulting 监管事务副总裁 Greg Meyer 表示,这种加快的步伐可部分归因于不断发展的制造技术。周转时间令人震惊,您可以非常快速地完成大量开发工作。 因此,虽然拥有大量员工的新超级办公室是一个良好的开端,但从长远来看可能是不够的。Majors 说,细胞和基因治疗 (CGT) 框架的监管方面以及准入和报销方面需要进行现代化改造,以确保这些疗法能够惠及患者。 “OTP 的创建及其向超级办公室的转变向我们发出了信号,表明 FDA 正在采取这些步骤实现现代化,并且 它将细胞和基因疗法视为未来医学领域极其重要的一部分。” Majors 和其他人强调了他们认为现代化应该解决的其他关注领域:标准化、化学、制造和控制 (CMC)、协作和专业知识。 “快速将安全有效的细胞和基因疗法推向市场以治愈尽可能多的这些疾病将需要在开发过程的每个步骤中利用效率、最大化标准化和充分利用现有数据的监管框架,”Karin国家罕见疾病组织政策和监管事务主管表示。 同时,ASGCT CEO David Barrett强调了对最初为小分子药物设计的CMC框架进行适配的必要性。 CGT 产品的开发是不同的,它们的最终 CMC 数据通常出现在产品生命周期的后期。 Ndugga-Kabuye 赞同 Hoelzer 和 Barrett 的说法,即 CMC 考虑因素的标准化应该是重中之重。 同时担任 CBER 副主任的 Witten 表示,FDA 已经意识到这些担忧,并“坚定地致力于与利益相关者合作……。. . 促进细胞和基因治疗领域的有益创新,以帮助提高这些创新产品的开发和批准效率。” 不过,她补充说,目前提交给 OTP 的监管审查程序没有变化。“应用程序将继续在当前流程中进行路由和审查。” 事实上,专家们承认,这种转变需要时间。监管机构和开发商都在不断学习,因为我们以前从未这样做过。 但这些改变是必要的,巴雷特补充道。“对于 CGT 研究来说,这是一个真正令人惊叹的时代,我们需要与科学进步同步应对监管挑战。患者需要新的模型才能获得挽救生命的治疗。”
编辑:小果果,转载请注明出处:https://www.cells88.com/zixun/hydt/32585.html
免责声明:本站所转载文章来源于其他平台,主要目的在于分享行业相关知识,传递当前最新资讯。图片、文章版权均属于原作者所有,如有侵权,请及时告知,我们会在24小时内删除相关信息。
说明:本站所发布的案例均摘录于文献,仅用于科普干细胞与再生医学相关知识,不作为医疗建议。
 微信扫一扫
微信扫一扫  支付宝扫一扫
支付宝扫一扫 