

介绍
随着嵌合抗原受体(CAR) T细胞的近期发展和出现,过继性细胞疗法发生了革命性变化。这些是经过改造的T细胞,在细胞膜上具有单链可变片段(scFv),可识别肿瘤抗原并能促进抗肿瘤活性。典型的第一代CAR构建体由融合跨膜结构域和细胞内信号单元(CD3 zeta链)的scFv组成,可实现肿瘤特异性表位识别和T细胞活化,而不依赖于主要组织相容性复合物。
该技术的发展为血液系统恶性肿瘤的治疗产生了持续的抗肿瘤反应和前所未有的结果。随着抗CD19 CAR-T细胞的初步临床成功,以及KYMRIAH™ (tisagenlecleucel)和YESCARTA™ (axicabtagene ciloleucel) 的监管批准),CAR-T 细胞的临床管线已大幅增加,目前在不同的临床试验中正在研究≥90种 CAR-T细胞治疗候选药物。尽管最初的临床研究仅限于血液系统恶性肿瘤,但该领域正在迅速向潜在发展方向发展用于实体瘤靶点。
CAR对肿瘤抗原的识别和参与导致CAR-T细胞的激活、肿瘤细胞的细胞溶解和细胞因子的释放。细胞因子随后促进了CAR-T细胞的快速扩增,随后促进了它们的记忆分化。尽管对CAR-T细胞的作用机制已经有了普遍的了解,但潜在的关键决定因素对影响其速率和程度的定量影响CAR-T细胞活性仍然知之甚少。
CAR-T细胞的药代动力学-药效学(PK-PD)关系的表征由于其在体内的自我复制和长期持续能力而呈现出许多挑战和独特的机遇。CAR-T细胞的典型多相处置概况包括快速分布阶段导致有时间限制的扩张阶段,然后是收缩和延长的持续阶段。尽管最近已经使用数学模型来表征CAR-T细胞的不同PK曲线,但无法利用经验模型来了解药物和系统特定参数如何促成这种独特的PK行为。因此,开发基于机制的转化PK-PD 模型,将关键的药物特异性和系统特异性参数整合到一个定量框架中,对于理解CAR-T细胞的关键PK-PD决定因素是非常宝贵的。
这样的模型可以:(1)促进先导 CAR 结构的设计和开发,(2)在临床前环境中对先导CAR-T候选物进行分类,以及(3)实现有效的临床前到临床转化。
在这里,我们采用逐步方法开发多尺度、机械PK-PD模型,使用针对多种CAR结构报告的一组综合文献数据,定量描述体外和体内临床前模型中的CAR-T细胞活性。
在第1步中,开发了细胞级PD模型以定量表征药物特异性(例如,CAR亲和力和 CAR密度)和系统特异性(例如,抗原密度、肿瘤负荷)参数对体外的影响CAR-T细胞活动,包括肿瘤细胞耗竭、CAR-T细胞扩增和细胞因子释放。
在第2步中,开发了一个基于生理的药代动力学(PBPK)模型来表征 CAR-T细胞在异种移植小鼠模型中的体内生物分布。
最后,在步骤3中,建立了PBPK-PD模型,以同时表征异种移植小鼠模型中的体内CAR-T扩增和肿瘤细胞耗竭。然后将体外效力与体内估计值进行比较,以建立体外和体内相关性 (IVIVC)。开发的PBPK-PD模型用于进行模拟,以了解CAR-T细胞PK-PD在CAR-T剂量水平和肿瘤负荷变化时的行为。我们在此提出的转化模型有望提供更好的框架来解释未来CAR-T细胞的临床PK-PD行为。
结果
用于表征体外CAR-T细胞活性的细胞水平PD模型
开发了一个数学模型(图 1a,在方法部分中描述)来表征CAR结合亲和力、靶细胞上的抗原表达、T细胞上的CAR表达和不同效应细胞:靶细胞(E:T)与同时定量描述体外靶细胞耗竭、细胞因子释放和T细胞扩增。为了开发这个体外模型,使用了一个综合数据集,包括两种不同的CAR结构,即抗表皮生长因子受体(EGFR) 和抗人表皮生长因子受体 2 (HER2) CAR-T细胞(在表格1)。使用该模型表征的三个定量结果包括:(1) 靶细胞耗竭,(2) CAR-T细胞增殖,和(3)细胞因子(例如干扰素(IFN)-γ)的释放。
表 1. 用于开发所提出的转化PK-PD模型的临床前体外和体内数据集
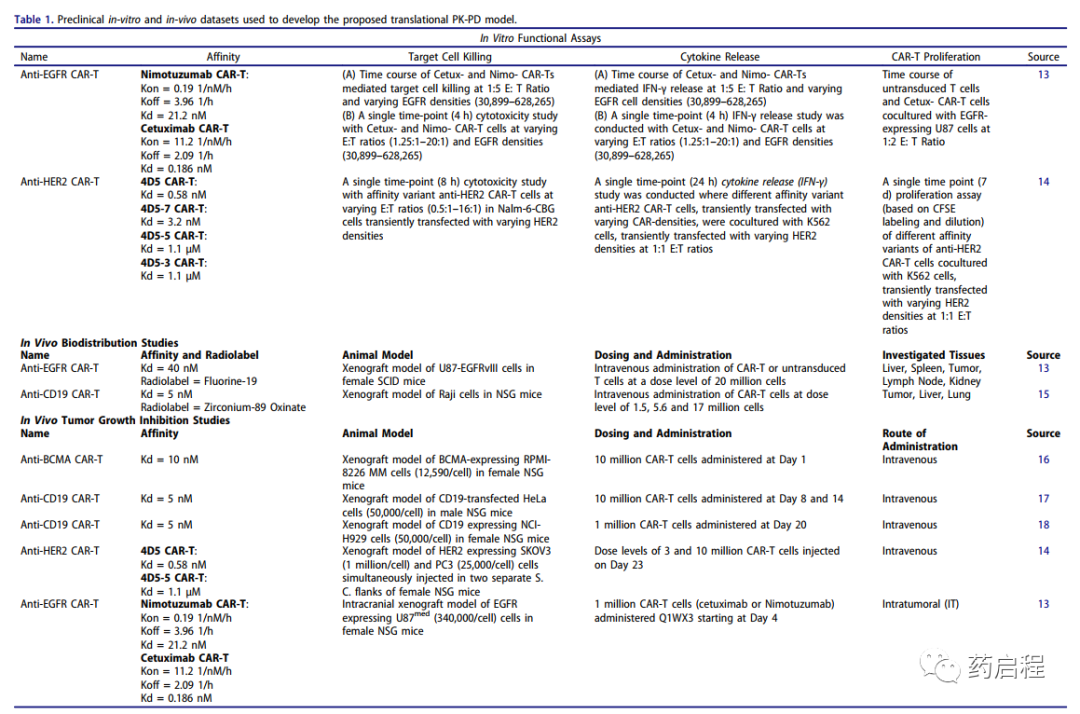

图 1. (A) CAR-T细胞活性的细胞级药效学模型示意图
抗EGFR CAR-T细胞
细胞水平模型用于拟合Caruso等人报道的抗EGFR CAR-T细胞的体外数据集。图 2描述了观察到的数据集和模型拟合曲线,用于表达EGFR的U87肿瘤细胞系耗竭(图 2a ),抗EGFR CAR-T细胞增殖(图2b)和细胞因子释放百分比相对于基线水平(图2c)在具有不同抗原密度(30,899-628,265个受体/细胞)的EGFR+U87细胞系的体外系统中)、CAR亲和力(西妥昔单抗与尼妥珠单抗 CAR-T)和E:T比率(1.25:1–20:1)(详细描述在表1中)。
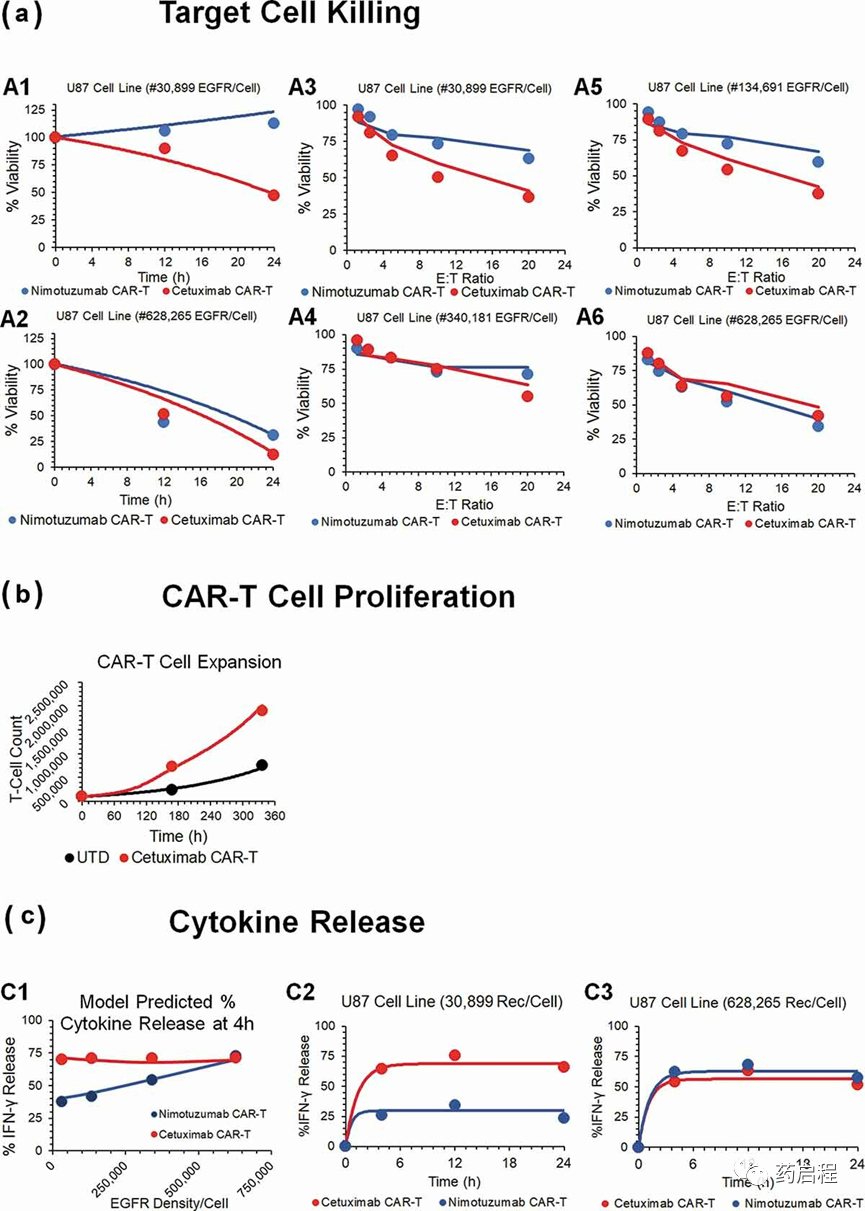
图 2. 体外系统中亲和力变体抗EGFR CAR-T细胞活性的观察和模型拟合曲线。
与高亲和力西妥昔单抗 CAR-T(红色轮廓)相比,该模型能够同时捕获低亲和力Nimotuzumab CAR-T(蓝色轮廓)在具有不同肿瘤细胞的 U87 细胞系中的不同肿瘤细胞杀伤潜力。EGFR密度,通过估计关键功效参数。该模型使用“CAR-靶标复合物/肿瘤细胞数量”的形成,由于更高的靶标参与度,这将随着亲和力和抗原密度的增加而增加。估计的效力参数值(KCCAR-T50,表 2)表明,需要约1.45个CAR-Target 复合物/肿瘤细胞来诱导CAR-T细胞表现出的最大杀伤率的50%。
表 2.用于构建CAR-T的PBPK-PD模型的固定或估计参数
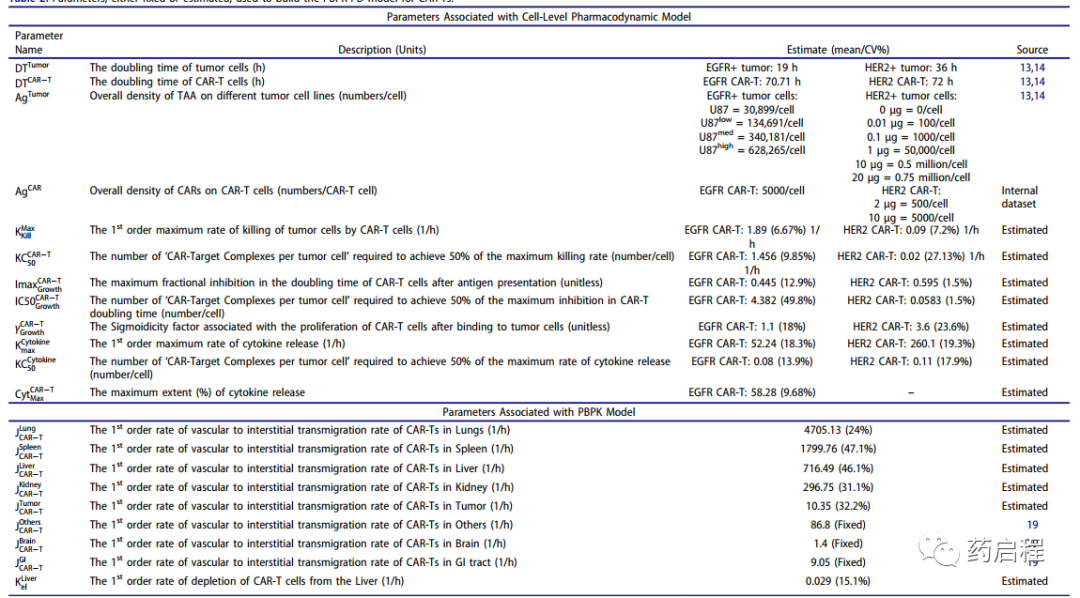

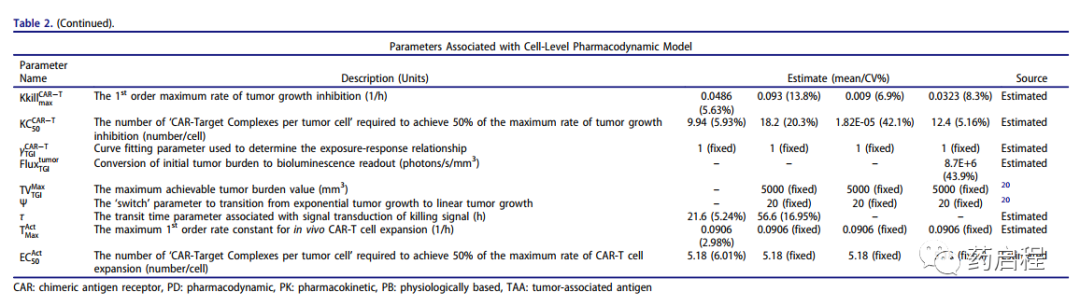
图 2b突出显示了与表达EGFR的U87细胞系共培养后,与未转导 (CAR-) T细胞相比,西妥昔单抗CAR+ T细胞的扩增。当与抗原阳性肿瘤细胞共培养时,该模型能够捕获激活诱导的CAR+ T细胞增强的增殖潜力。根据等式4和11中描述的结构模型,估计的参数(表 2)表明每个肿瘤细胞需要约4.3个CAR-Target复合物才能使CAR-T细胞加倍最大减少50%时间,而在所研究的体外系统中,CAR-T细胞倍增时间(ImaxCAR-TGrowth)的最大可饱和减少估计为44.5%。
图 2c描绘了在CAR-T细胞的两种不同亲和力变体与不同EGFR表达细胞系相互作用时,与观察到的数据叠加的模型拟合,用于IFN-γ 的百分比增加(与基线相比)。该模型能够同时表征一个综合数据集,以作为时间、EGFR受体密度和CAR亲和力的函数,在体外共培养中 IFN-γ 的饱和增加。相关的模型参数(方程12,表 2)反映了在所研究的体外系统中,~0.1 CAR-Target 复合物/肿瘤细胞能够诱导50%的最大激活诱导细胞因子释放率。
抗HER2 CAR-T细胞
细胞水平PD模型(图 1a)还用于表征Liu等人报道的亲和力变异抗HER2 CAR-T细胞的体外数据集。图3描述了观察到的 HER2 数据集和模型拟合曲线-在综合体外实验中表达肿瘤细胞耗竭(图 3a)、抗HER2 CAR-T细胞增殖(图 3b)和诱导绝对浓度的IFN-γ(以 pg/mL 为单位,图 3c) CAR亲和力、HER2受体密度、CAR密度和E:T比率(在表 1 中详细描述)。
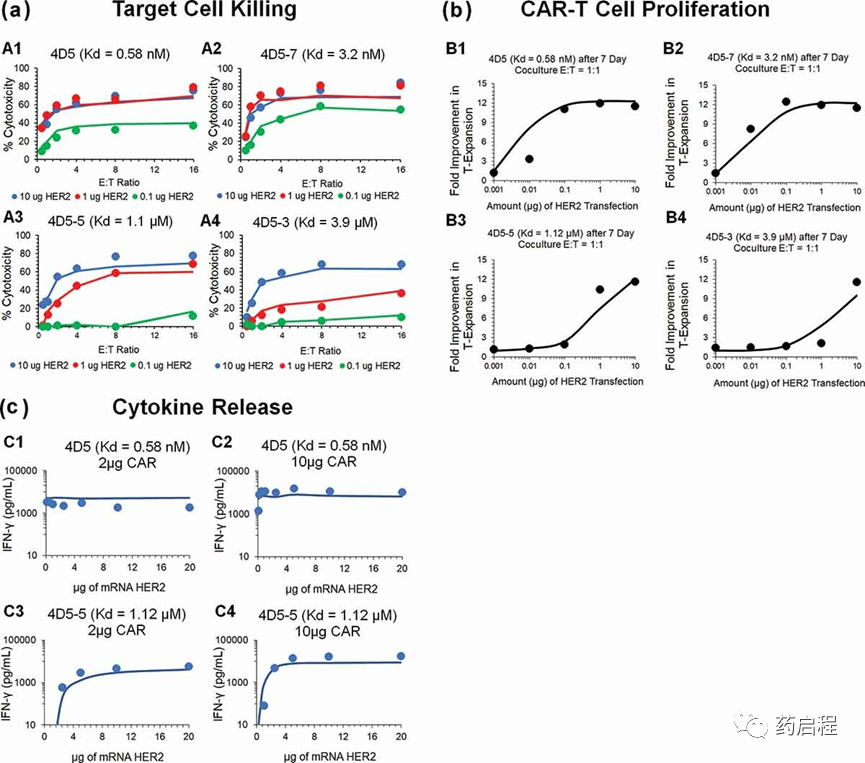
图 3.体外系统中亲和力变体抗HER2 CAR-T细胞活性的观察和模型拟合曲线。
图 3A描绘了与8小时体外细胞毒性实验的观察数据叠加的同步模型拟合曲线,其中四种不同的亲和力变体(Kd范围为3.9µM–0.58nM)的 CAR-T细胞与Nalm-6-CBG共培养细胞系,转染不同水平的HER2 mRNA。该模型能够考虑不同的系统特异性决定因素(例如,CAR-亲和力、HER2密度)以有效捕获可饱和杀伤曲线,并估计一组通用的体外功效参数。模型估计的效力(KCCAR-T50,表 2)表明,需要约0.05 个 CAR-Target复合物/肿瘤细胞才能诱导 CAR-T 细胞表现出的最大杀伤率的50%。
图 3b 描述了与未转导的T细胞相比,当与具有不同抗原密度的肿瘤细胞共培养7h后,与未转导的T细胞相比,与观察到的数据集重叠的模型拟合曲线。数据集的同时表征能够识别在存在关键潜在决定因素(例如 CAR 亲和力和抗原密度)的情况下 CAR-T 细胞扩增的速率和饱和程度。参数估计(表 2,等式4 和 11)显示,需要 0.06 CAR-Target Complexes/肿瘤细胞来实现 CAR-T细胞倍增时间的最大减少 50%,而最大可饱和在体外系统中,CAR-T细胞倍增时间 (ImaxCAR-TGrowth) 的抑制率估计为 59.5%。
图 3c描述了与K562细胞共培养后的模型拟合曲线,与观察到的产生IFN-γ(绝对浓度,pg/mL)的数据重叠,用不同水平的 HER2 瞬时转染,具有亲和力变体抗 HER2用不同数量的CAR mRNA转导的CAR-T细胞(详见表 1)。所提出的模型结构能够使用估计的CAR-Target 复合物 (CplxCell) 的数量来同时表征不同数据集中细胞因子释放的速率和程度。建模结果显示,每个肿瘤细胞需要约 0.1个CAR-Target复合物才能达到50%的最大细胞因子释放率。此外,与抗EGFR案例研究一致,在所研究的体外系统中估计了细胞因子释放的非常快的最大速率 (KMaxCytokine)。
基于生理学的PK模型来表征CAR-T细胞的生物分布
开发了一个 PBPK 模型来表征异种移植小鼠模型中CAR-T细胞的PK和组织分布。开发的模型用于表征来自抗EGFR CAR-T细胞和抗CD19 CAR-T细胞的生物分布数据集。有关CAR结构、CAR亲和力、静脉内(IV)剂量水平、使用和研究的放射性标记的详细信息与每项研究相关的组织列于表1。模型结构的示意图如图1b所示,详细的模型描述在方法部分提供。
图 4 描述了与非转导(CAR-) T细胞 (图 4a)、抗EGFR CAR+ T细胞 (图 4b) 和抗CD19 CAR+ T的生物分布数据集同时拟合相关的模型拟合曲线和观察到的数据点异种移植小鼠模型中的细胞(图 4c)。所提出的 PBPK 模型同时表征了所有三种CAR构建体的生物分布,以及所有主要器官的一组血管到间质迁移 (JOrgan) 率。在进行模型拟合时,与每个案例研究相关的生理参数(例如,流速、器官体积)以及药物和系统特定参数(例如,CAR亲和力、肿瘤体积、抗原密度),固定为文献报道的值,如表2和补充表1所列。在用于PBPK的数据集(抗EGFR和抗CD19)中未观察到肿瘤分布和抗原介导的刺激后CAR-T细胞的扩增模型构建,大概是由于研究的时间点有限,因此没有包含在模型结构中。然而,CAR-T 细胞扩增后来在开发PBPK-PD关系时实施。
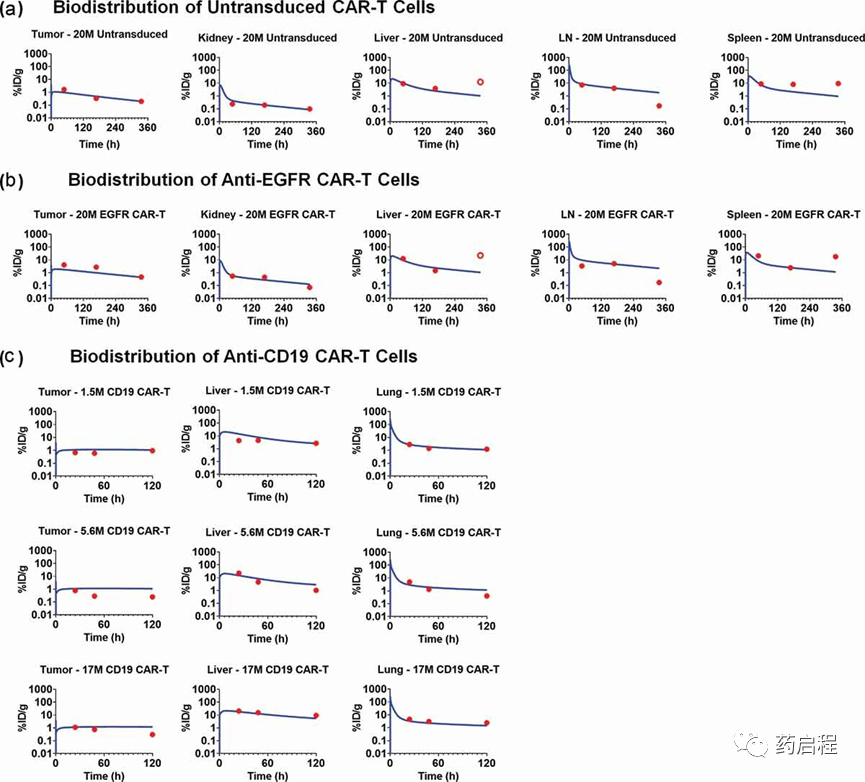
图 4. CAR-T 细胞在异种移植小鼠模型中的生物分布的观察和模型拟合曲线。
与提议的 PBPK 模型相关的估计参数包括所有组织的CAR-T细胞生物分布可用的一阶迁移率(JOrgan,如参考文献19,22中所述)(表 2)。对于没有生物分布数据的其他组织(例如,脑、胃肠道[GI]道、其他),迁移率(JOrgan)固定为 Khot 等人报告的值。
此外,一阶消除CAR-T细胞(KLiverel)的比率是根据肝室的血管外空间 (VEVLiver) 估计的,基于多个关于肝脏消除 CAR-T 细胞的报告。由于模型估计的迁移率(Jorgan)是反映不同组织之间的分布率,观察到T细胞的最大分布在肺、脾和肝中。模型估计的 CAR-T 细胞从肝脏血管外VEVLiver空间中的消除率为0.029 1/h(~ 1-d 半衰期)。
图4a和4b代表了携带U87异种移植小鼠模型的研究组织中未转导T细胞和抗EGFR CAR-T细胞(CAR亲和力:Kd为40nM)的观察数据和模型拟合图谱。虽然提出的模型解释了CAR-Target复合物的形成,与未转导的T细胞(图 4a)相比,模型预测的CAR-T细胞(图 4b)在肿瘤内的生物分布曲线没有明显差异。可能的原因之一可能是因为 CAR-T 扩展在靶标参与肿瘤后没有合并。值得注意的是,三个案例研究的%ID/g非常一致(比较图4a-c时)。此外,根据抗CD19 CAR-T的生物分布结果,其中施用了三种不同剂量(即1.5、5.6和1700万个CAR+细胞),观察到CAR-T细胞水平的动力学约为剂量-在剂量范围内成正比。因此,从肝脏血管外空间 ((VELViver)) 消除的一阶线性速率被用于表征CAR-T细胞的分布。
开发PBPK-PD模型来表征异种移植小鼠模型中的体内CAR-T细胞扩增和肿瘤生长抑制
将开发的PBPK模型扩展为具有PD建模组件,该组件使用“每个肿瘤细胞的 CAR-Target复合物数量”作为肿瘤生长抑制(TGI)和CAR-T细胞扩增的驱动因素。模型结构的示意图如图1c所示,详细的模型描述在方法部分提供。
抗BCMA CAR-T (bb2121)细胞
图 5a描述了在单次IV施用5×106抗BCMA(bb2121) CAR+ T细胞后,表达B细胞成熟抗原 (BCMA) 的RPMI-8226异种移植小鼠中TGI和 CAR-T细胞扩增的同时表征/老鼠。如方法部分中详细描述的,动态(具有生长和杀伤功能)肿瘤隔室(图 1C)被纳入PBPK模型,其中“每个肿瘤细胞的CAR-Target复合物数量”用于驱动功效和扩增肿瘤血管外(VEVTumor)空间中的总CAR-T细胞(未结合和肿瘤细胞结合)。与之前描述的用于PBPK模型开发的生物分布研究相比,随着该PK-PD研究的持续时间更长和采样频率更高(长达28天),观察到CAR-T细胞在血液隔室中扩增(图 5A)。
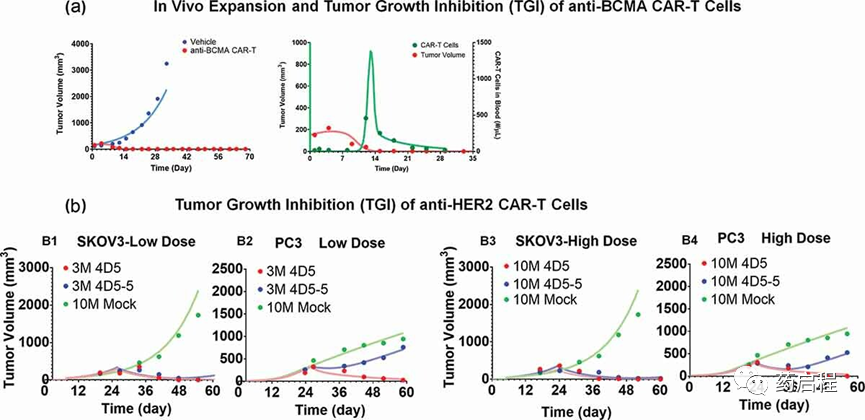
图 5.CAR-T细胞体内扩增和CAR-T诱导的肿瘤生长抑制的观察和模型拟合曲线。
在血液中静脉注射CAR+ T细胞后,早期未观察到血液中可检测到的CAR-T细胞水平,突出了快速分布/边缘阶段。该模型能够捕获这种独特的配置文件,如图5A所示(绿色配置文件)。在与肿瘤血管外空间(VEVTumor)内的肿瘤细胞相互作用后,CAR-T细胞会扩增,最终循环回(通过淋巴流)到血液中(绿色轮廓,图 5A),如PBPK模型所预测的那样在图1B。这种现象可能导致TGI诱导的明显延迟(红色轮廓,图5A)和血液隔室中CAR-T细胞的明显扩张(绿色轮廓,图5A)。肿瘤细胞在第14天死亡后,“CAR-Target Complex”驱动信号减少,导致快速收缩阶段,此时CAR-T细胞迅速下降,最终符合其基线暴露水平。
有趣的是,模型估计的肿瘤细胞最大杀伤率 (KKillMax) (~0.05 1/h) 和“每个肿瘤细胞的 CAR-Target 复合物数量”(KCCAR-T50)需要 (9.94个复合物/肿瘤细胞) 50%的最大肿瘤耗竭率与抗 HER2 和抗 EGFR CAR-T 细胞的体外估计值相似(表 2)。诱导活化CAR-T细胞扩增所需的模型估计最大速率 (TActMax) 和“每个肿瘤细胞的CAR-Target复合物数量”(ECAct50)分别为0.09 (1/h)和5.18 (复合物/肿瘤细胞)。体内观察到的T细胞扩增效力估计值(ECAct50)也与之前的体外效力估计值(ICCAR-TGrowth)非常相似,突出了在开发转化PK-PD关系时采用系统方法的潜在效用。
抗HER2 CAR-T细胞
图5B描述了在皮下接种 HER2-high SKOV3 的异种移植小鼠中的亲和力变异(4D5[高亲和力] 和4D5-5[低亲和力])抗HER2 CAR-T细胞的TGI曲线(观察和模型拟合)使用图1C中描述的模型(参见方法部分)。使用提出的PBPK-PD模型对所有数据集(低剂量和高剂量)进行同时表征。所有药物特异性(CAR亲和力和CAR密度)和系统特异性(SKOV3和PC3的抗原密度)参数都固定为已知值,而与肿瘤生长和杀伤相关的参数是估计的(表 2)。由于当前案例研究中缺乏此类数据,与 CAR-T 细胞体内扩增相关的参数被固定为抗 BCMA(bb2121)研究的估计值。
该模型同时表征HER2-high SKOV3和HER2-low PC3 TGI中的TGI的能力支持了 CAR亲和力、靶标丰度和CAR-T细胞活性之间的定量关系可以转化为体内环境,其中与低表达PC3肿瘤相比,低亲和力和高亲和力CAR-T细胞在高表达SKOV3肿瘤中的相对疗效不同。由于4D5-5(蓝色轮廓)的结合亲和力较低,与高HER2 SKOV3异种移植物相比,预计低HER2 PC3异种移植物中的“每个肿瘤细胞的CAR-Target Complexes”形成较少,最终导致在TGI的分化中。
抗CD19 CAR-T细胞
图 6表示在接种了表达CD19的HeLa或H929细胞的异种移植小鼠模型中的两个不同TGI实验的汇总观察数据和模型拟合曲线。表1中描述了两种细胞系的受体密度和给药方案的更多详细信息。所提出的PBPK-PD模型(图 1C)能够同时捕获用磷酸盐缓冲盐水(PBS)对照处理的动物的肿瘤生长曲线、未转导(CAR-) T细胞和抗CD19 CAR-T 细胞,在单次和多次给药方案下。由于本研究中缺乏CAR-T细胞计数,与“CAR-Target complex”驱动的CAR-T细胞体内扩增相关的参数固定为从抗 BCMA (bb2121) 案例估计的值研究(表 2)。该模型能够同时表征来自不同研究、异种移植模型和给药方案的汇总数据集。模型估计的体内肿瘤细胞消耗的最大速率 (KKillmax)和效力(KCCAR-T50)分别估计为0.093(1/h)和18.2(CAR-Target复合物/肿瘤细胞的数量)(列于表 2)。
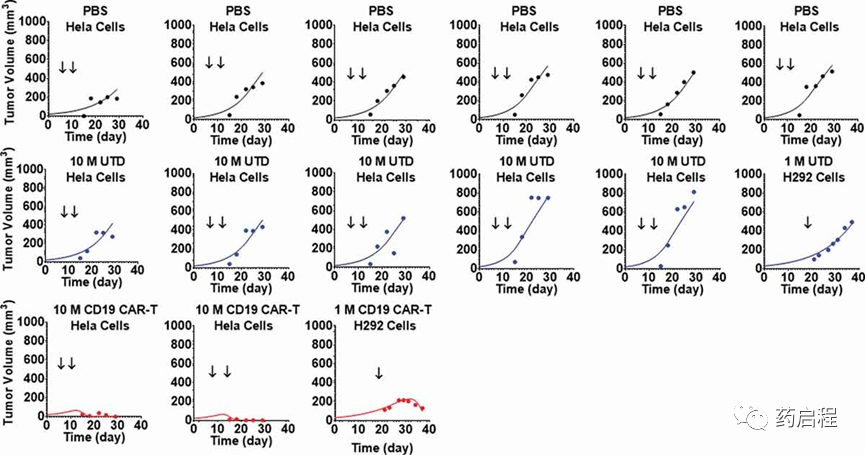
图 6. 抗CD19 CAR-T诱导的肿瘤生长抑制的观察和个体模型拟合曲线。
抗EGFR CAR-T细胞
图 7描述了在胶质母细胞瘤原位小鼠模型中观察到的数据集和模型拟合的TGI 曲线,接种了表达EGFR的U87细胞。用PBS对照或1×106个高亲和力西妥昔单抗 CAR-T或低亲和力尼妥珠单抗CAR-T的CAR+细胞治疗动物,通过瘤内(IT)注射以Q1WX3给药方案给药。在开发的PBPK模型中,CAR-T细胞的推注剂量被描述为在肿瘤隔室的血管外空间内给药(图 1B)。使用PBPK-PD模型同时表征TGI谱使得能够表征由于EGFR结合亲和力对CAR-T细胞的影响而导致的CAR-T细胞的不同功效,证实了 EGFR结合亲和力对CAR-T细胞的定量影响在体内环境中的活动。模型估计的体内肿瘤细胞消耗的最大速率(KKillmax)和效力(KCCAR-T50)分别估计为0.032 (1/h) 和 12.4 (CAR-Target 复合物/肿瘤细胞的数量) (列出在表2中)。
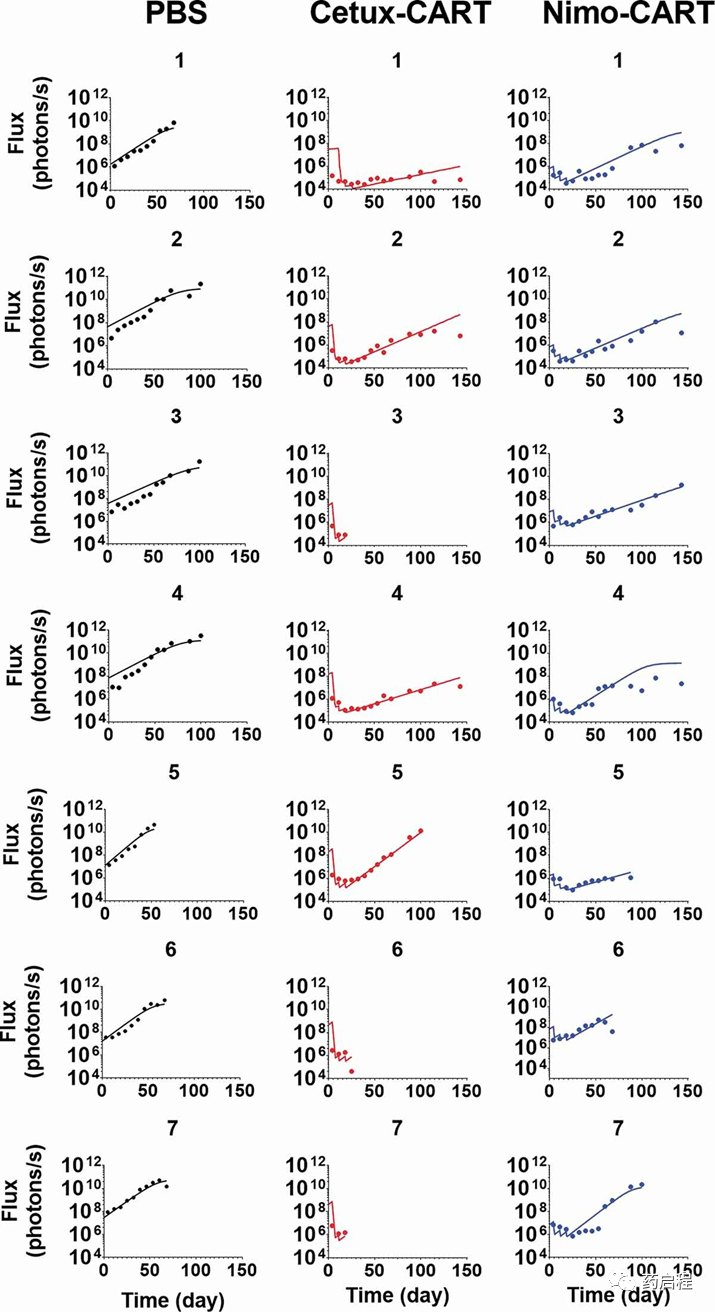
图 7. 抗EGFR CAR-T 诱导的肿瘤生长抑制的观察和个体模型拟合曲线。用表达 EGFR 的 U87 细胞(第 0 天)接种的颅内异种移植小鼠接受瘤内(IT)给药(1)
基于模型的模拟研究剂量和肿瘤负荷对肿瘤动力学、靶标参与和CAR-T细胞扩增的影响
为了进一步评估我们开发的PBPK-PD模型的体内相关性,进行了基于模型的模拟以检查剂量和肿瘤负荷的影响,并将结果与临床观察到的结果进行比较。图 8描述了TGI曲线(图8A和8D)、肿瘤血管外空间 (VEVTumor) 中“每个肿瘤细胞的 CAR-Target Complexes 数量”(图 8B 和 8E)以及CAR-T浓度的模型模拟基于bb2121案例研究的模型估计参数。如下所述,模拟结果揭示了一些有趣的趋势。
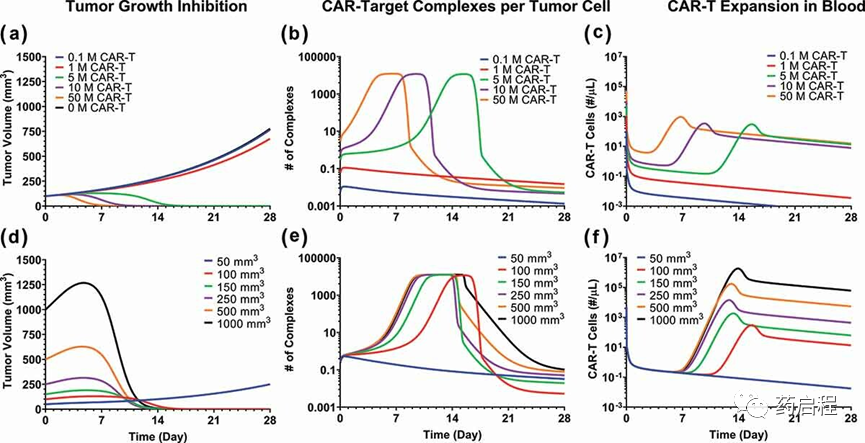
图 8. 使用经过验证的 PBPK-PD 模型进行模型预测,以同时评估 (1) CAR-T 剂量和 (2) 初始肿瘤负荷对 (A 和 D) 肿瘤生长抑制 (TGI)、(B 和 E) 生成的影响肿瘤血管外空间中“每个肿瘤细胞的 CAR-Target 复合物数量”和(D 和 F)CAR-T 细胞在血液中的扩增:1)CAR-T 剂量:在单次静脉注射抗 BCMA 后进行模拟( bb2121)携带异种移植物的 RPMI-8226 中的 CAR-T 细胞,剂量水平为每只小鼠 0.1、1、5、10 和 5000 万个 CAR-T 细胞。(2) 初始肿瘤负担:在初始肿瘤负担为 50 , 100, 150, 250, 500 和 1000 mm3。
剂量的影响
模拟结果表明,与对照(黑色曲线)相比,在CAR-T达到临界剂量水平之前,没有观察到显着的TGI。在这个临界剂量水平之外,有一系列剂量具有陡峭的剂量-暴露关系,然后剂量对暴露的影响趋于稳定(图 8A)。结果与模拟的“每个肿瘤细胞的CAR-Target复合物数量”一致(图 8B),即在低剂量水平(红色和蓝色曲线)下,每个肿瘤细胞几乎没有形成 CAR-靶标复合物.然而,在更高的剂量水平下,CAR-Target复合物(ECExp50=5.18 复合物/肿瘤细胞)的数量迅速增加,这会同时启动肿瘤杀伤(图 8A)和 CAR-T 细胞的扩增(图 8C)。随着剂量水平的进一步增加,CAR-Target 复合物的形成速度(图 8B)以及TGI的速度(图 8A)增加。这种现象导致肿瘤负荷迅速下降(图 8A),因此抗原丰度总体下降。随着肿瘤负荷的下降,由于肿瘤空间内没有抗原相互作用,CAR-Target Complexes 的总数也迅速下降,导致CAR-T增殖减少。
图 8C说明了血液隔室内的预期CAR-T动力学。在较低剂量水平下,PK曲线仅限于(蓝色和红色曲线)快速初始分布(到组织),然后是持续消除阶段(图 8C)。然而,在更高的剂量水平下,预计CAR-T细胞会出现剂量依赖性的扩增率。模拟表明,预计CAR-T细胞扩增的总体程度(Cmax)不会超过某些CAR-T剂量水平,而达到Cmax的时间(Tmax)可能会随着剂量的增加而减少。之后,在抗原耗尽后 CAR-Target Complexes快速减少后,CAR-T细胞的血液PK出现快速收缩阶段,随后是一级消除的持续阶段。
肿瘤负荷的影响
还评估了基线肿瘤负荷对肿瘤动力学(图 8D)、每个肿瘤细胞形成CAR-Target复合物(图 8E)和血液中CAR-T细胞扩增(图 8F)的潜在影响。有趣的是,模型模拟表明,尽管初始肿瘤负荷存在差异,但CAR-T细胞可以在相似的时间范围内实现肿瘤消除。这是因为更高的肿瘤负荷导致“每个肿瘤细胞的CAR-Target复合物”(图 8E)更快但饱和的形成,这固有地导致CAR-T扩展的速率(较低的Tmax)和程度(Cmax)增加(图 8F)和更快地杀死肿瘤细胞 (图 8D)。
CAR-T细胞PBPK-PD模型的全局敏感性分析
图 9描述了针对抗BCMA(BB2121)CAR-T细胞(图 5A,表 2)开发的 PBPK-PD 模型对血液 CAR-T 细胞扩增(图 9A)和整体肿瘤的全局敏感性分析(GSA)体积(图 9B)。我们观察到与“CAR-Target 复合物”形成相关的参数主要对CAR-T细胞的扩增阶段敏感(图 9A)。CAR-affinity(Kon和Koff)、CAR密度和抗原密度等参数与扩增程度呈正相关。GSA关于初始肿瘤负荷和剂量对CAR-T细胞扩增的影响的结果与我们之前的结果非常相似,如图8所示,其中较高的肿瘤负荷导致较高的 Cmax,而较高的CAR-T剂量导致较短的Tmax。
图 9B描述了不同参数对肿瘤体积的敏感性。导致更高“CAR-Target复合物”形成的大多数参数(例如CAR-亲和力、CAR-密度和抗原密度)与总体肿瘤体积呈负相关。
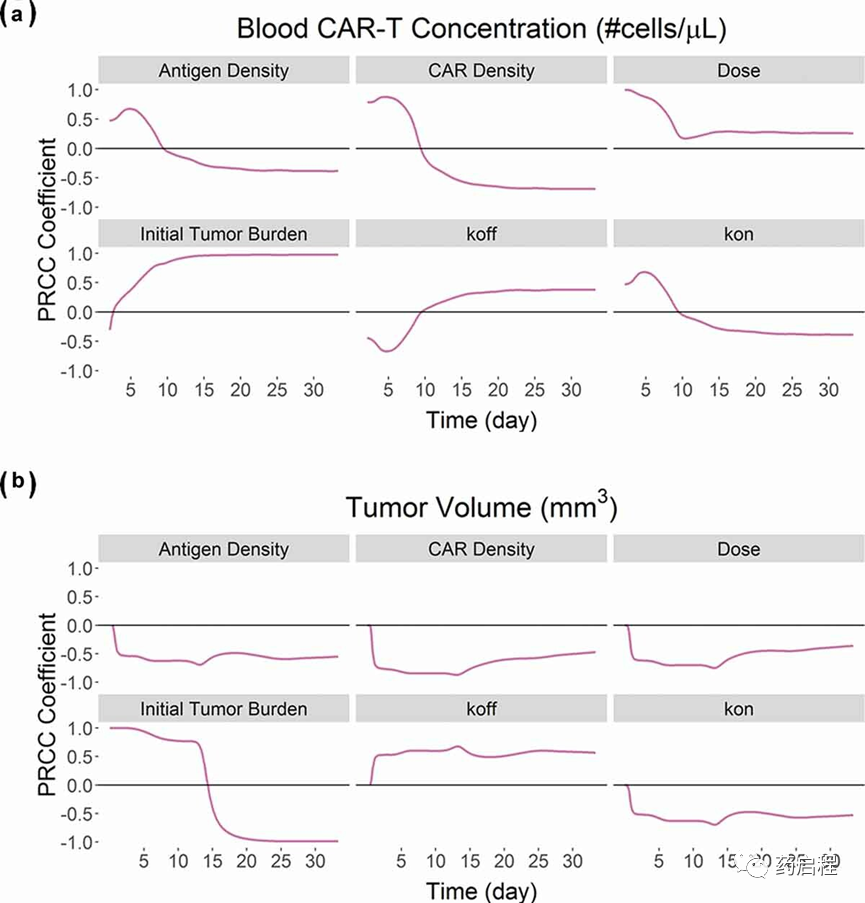
图 9. BB2121已开发的PBPK-PD模型的全局敏感性分析结果:基于PRCC的“抗原密度”、“CAR密度”、“CAR-T剂量”、“初始肿瘤负荷”、“Koff”的敏感性指标’ 和 ‘Kon’ 在 (A) 血液 CAR-T 浓度和 (B) 总肿瘤体积
讨论
用 CAR 转导的 T 细胞的过继细胞转移已经彻底改变了临床免疫治疗领域。这些“自我复制剂”在对血液系统恶性肿瘤患者给药后显示出显着的临床疗效和长期持久性。这些药物的有前景的属性已得到监管机构的认可,2017 年,两种 CAR-T 细胞疗法获得了美国食品和药物管理局的批准。尽管取得了巨大成功,但关键药物特异性和系统性的定量影响临床中与 CAR-T 细胞活性相关的特定决定因素尚不清楚。为 CAR-T 细胞建立 PK-PD 关系具有挑战性,并且没有既定的范式或指南来预测人体 CAR-T 细胞的安全有效剂量水平。开发多尺度系统 PK-PD 模型可能是确定与这些药物的动力学和活性相关的关键决定因素的非常有益的第一步。
Stein等人最近使用来自tisagenlecleucel(抗 CD19)临床试验的2期数据集提交了第一份关于CAR-T细胞临床PK表征的报告。该模型将血液循环中的CAR-T细胞动力学分为三个不同的阶段:(1)初始时间受限的指数扩张阶段,随后是(2)快速收缩阶段,然后是(3)持续持续阶段。该模型成功地捕获了人类独特的CAR-T动力学曲线,并估计了CAR-T动力学每个不同阶段的斜率。然而,该模型的描述性限制了对其他CAR-T细胞疗法和替代剂量水平的外推。
最近,Hardiansyah等人采用了一种更系统的方法来表征临床CAR-T细胞PK和细胞因子释放动力学,他们利用肿瘤动力学来驱动CAR-T细胞扩增以及效应器和记忆 T 细胞之间的相互转换表型。该模型对不同的CAR-T细胞分布动力学的潜在机制提供了一些见解,并突出了CAR-T动力学和PD的综合性质。尽管他们最初的数据分析仅限于两名患者,在参数估计中观察到较大的受试者间变异性,但它证明了使用基于机制的模型来捕获CAR-T细胞动力学的可行性,即时间-依赖扩张,快速收缩和持久的持久性。
在这里,我们使用了一种基于机制的自下而上的方法来量化与CAR-T细胞活性和独特的CAR-T细胞PK行为相关的关键药物和系统特定参数的影响。我们建模方法的第一步涉及开发细胞级PD模型(图 1A),该模型考虑了CAR亲和力、CAR密度、抗原密度和E:T比率的影响,以计算“每个肿瘤细胞的CAR靶标复合物,因此决定了可饱和肿瘤细胞杀伤、CAR-T 扩增和细胞因子释放的速率和程度。使用来自一组综合体外实验的报告数据(如表 1 所述),该模型解释了动态E:T比率(由于T细胞扩增)、关键药物特异性(例如,CAR-亲和力和 CAR-密度)以及系统特异性(例如抗原密度)决定因素,同时估计关键效力参数。开发的细胞水平模型被证明可以定量捕捉一些药物特异性参数(如CAR亲和力和CAR密度)对体外整体CAR-T细胞活性的影响。
事实上,最近对CD19 CAR-T、CAT19的临床观察表明,与tisagenlecleucel (FMC63)相比,它具有较低的亲和力(更快的Koff率),因此具有更好的体外流动性,已证明具有更好的耐受性(即更低的在CARPALL临床试验中,与tisagenlecleucel (KYMRIAH™) 相比,同时保留了临床疗效的程度。在为 CAR-T 细胞开发转化体内PK-PD关系时,此类临床观察进一步强化了我们整合细胞水平信息的方法。我们设想未来将使用这种细胞级模型来识别最佳CAR-T 特征(即 CAR-亲和力和 CAR-密度),并促进在发现阶段选择领先的CAR-T候选者。
我们建模分析的第二步是开发一个PBPK模型来表征CAR-T细胞的全身分布。为 CAR-T开发PBPK模型至关重要,因为许多文献报道表明,预计总淋巴细胞群中只有一小部分(~2%)存在于外周血中。由于大多数抗原丰度(肿瘤对于开发中的 CAR-T 细胞的负载)通常位于骨髓、脾脏、淋巴结(血液学靶点)或实体瘤中,因此必须有一个强大的生理框架来表征血液:CAR-T 细胞的组织关系。
所提出的 PBPK 模型(图1B)结构基于Baxter等人和Khot 人发表的工作,用于表征CAR-T细胞的生物分布。该模型假设:(1)T细胞消除器官是肝脏,基于支持这一观点的多篇文献报道,和(2)CAR和肿瘤抗原仅在异种移植小鼠模型的肿瘤室内相互作用。PBPK模型能够同时表征所有主要感兴趣组织中未转导的 T 细胞、抗EGFR CAR-T细胞和抗CD19 CAR-T细胞的生物分布。脾脏、肝脏和肺被确定为 CAR-T 细胞生物分布最大的器官(当比较Jorgan值时),这与之前在异种移植小鼠和患者中的许多研究一致。
我们建模分析的第三步也是最后一步是建立一个整合的PBPK-PD关系,这同时解释了肿瘤结合和旁通未结合的CAR-T细胞的扩增,以及体内的抗肿瘤反应(图 1C)。该模型能够表征血液中抗 BCMA CAR-T 细胞的快速扩增阶段并观察到 TGI(图 5A)。该模型后来被用于表征 TGI 数据集:(1)亲和力变体抗 HER2 CAR-T(图 5B),(2)抗 CD19 CAR-T(图 6)和(3)抗 EGFR CAR -T(图 7)细胞。当在临床前环境中对先导 CAR 结构进行分类时,我们的转化建模框架可用于建立体外-体内相关性。基于不同 CAR 构建体的效力参数的总体比较(列于表 1),我们观察到体外效力值的等级排序可以转化为体内设置,并且体外效力值始终保持在 ~10 到 20比体内效力值高一倍(KCCAR-T50 较低,表 2)。与动态的体内情景相比,这一观察结果可能是由于 CAR-T 细胞在静态体外共培养环境中与肿瘤细胞接触的可能性更高。
最后,通过评估(1)剂量和(2)初始肿瘤负荷对CAR-Target复合物形成、抗肿瘤反应和CAR-T细胞在血液中扩增的影响,检查了开发的PBPK-PD模型的体内相关性(图 8)并将模拟与人类报告的CAR-T曲线进行比较。模型模拟表明,在形成阈值“每个肿瘤细胞的CAR-Target 复合物”后,CAR-T细胞会加速扩增(图 8C 和 8F),从而加速肿瘤细胞的根除(图 8A 和 8D) .在具有多个CAR-T计划(例如,抗CD19和抗BCMA CAR-T)的临床环境中观察到了类似的模式,其中在 CAR-T 细胞扩增时,在体内观察到快速的肿瘤耗竭2-3个月,完全响应率很高。血液 PK 的模型模拟(图 8C和8F)还显示,在静脉注射CAR-T细胞后,血液PK开始迅速下降,可能是由于分布到其他组织(快速分布阶段)。然而,一旦CAR-T细胞在作用部位(生物相)内扩增,它们就会再循环回血流中。在所有临床试验结果(例如tisagenlecleucel)中,血流中CAR-T细胞扩增阶段开始的延迟很普遍,尤其是在早期时间点(例如1-2个月)报告PK数据的情况下。模拟还表明,在总体肿瘤负荷耗尽后,CAR-Target复合物形成的数量减少(图 8B和8E),这导致CAR-T的血液 PK 内出现快速收缩阶段(图8C和8F),在剩余的 CAR-T 细胞进入一个延长的持续阶段之前,一级消除。这组模拟(图 8C 和 8F)与临床中 CAR-T 细胞动力学的典型趋势一致。
我们的模型模拟还表明了一个非常陡峭的 CAR-T 剂量-暴露关系(图 8C),然后是一个平台期。观察到的平台受限于“每个肿瘤细胞的CAR-Target复合物数量”的饱和形成(图 8B 和 8E)。这一观察结果与许多临床PK数据集一致,其中超过一定水平的剂量不再与观察到的 Cmax 相关。最近来自抗BCMA (bb2121) 1 期研究的临床PK数据集也揭示了陡峭的剂量暴露关系,其中超过阈值剂量(1.5 亿 CAR-T 细胞),所有随后的更高剂量水平产生非常相似的Cmax水平。在研究中,细胞因子释放综合征的发生也与暴露程度相关,因此高于阈值剂量。然而,扩展至 Cmax 的速率随着剂量水平的增加而增加(图 6B)。这在一些报道的抗 CD1936、37 和抗BCMA CAR-T细胞的临床PK 数据中观察到,其中注入更高剂量 CAR-T 细胞的患者实现更快的扩增至 Cmax(即更低的 Tmax)。模型模拟还揭示了患者肿瘤负荷的影响,这可能导致 CAR-T细胞暴露(包括 Cmax)的显着变化。这也与许多临床报告一致(针对抗CD19 CAR-T),其中较高的肿瘤负荷导致较高的 Cmax,因此更多的细胞因子释放。
总而言之,我们在这里描述了一种基于机制的模型,用于表征CAR-T细胞的 PK-PD。尽管需要进一步发展的模型来通过结合其他相关成分来描述 CAR-T 细胞的临床行为,例如CAR-T细胞记忆分化的动力学、CD4/CD8比率和细胞因子环境对体内扩增的影响CAR-T 细胞是我们的多尺度转化 PBPK-PD 模型,整合了药物和系统特异性参数,能够在体外和体内表征CAR-T细胞活性。与CAR-T细胞的不同亚群(CD4+与CD8+)和表型(干细胞记忆[Tscm]与效应T细胞[Te])相关的基于流式细胞术的测量的可用性将使我们的基础模型进一步发展,以解释差异扩张体内不同CAR-T细胞表型的能力和效力。尽管如此,我们目前的模型有望为理解 CAR-T 细胞的临床行为提供一个框架,从而促进未来 CAR-T 细胞疗法的设计和开发。


D
O
i
:
Development of a quantitative relationship between CAR-affinity, antigen abundance, tumor cell depletion and CAR-T cell expansion using a multiscale systems PK-PD model
https://doi.org/10.1080/19420862.2019.1688616
编辑:小果果,转载请注明出处:https://www.cells88.com/cells/myxb/9689.html
免责声明:本站所转载文章来源于其他平台,主要目的在于分享行业相关知识,传递当前最新资讯。图片、文章版权均属于原作者所有,如有侵权,请及时告知,我们会在24小时内删除相关信息。
说明:本站所发布的案例均摘录于文献,仅用于科普干细胞与再生医学相关知识,不作为医疗建议。
 微信扫一扫
微信扫一扫  支付宝扫一扫
支付宝扫一扫 