

近年来,以免疫检查点抑制剂(ICIs)为代表的免疫治疗在癌症治疗方面取得了前所未有的突破。然而,许多肿瘤对ICIs的反应很差甚至没有反应,部分原因是肿瘤浸润淋巴细胞(TIL)的缺乏,这大大限制了ICIs的应用。将这些免疫“冷”肿瘤转化为可能对ICIs产生反应的“热”肿瘤是癌症免疫治疗中尚未解决的问题。
由于抗凋亡是癌症的一个普遍特征,诱导非凋亡调节性细胞死亡(RCD)是一种新的癌症治疗策略。RCD在维持体内平衡和疾病发展中起着至关重要的作用。根据不同的形态学、生化、免疫学和遗传学特征,RCD可分为凋亡和非凋亡两类。非凋亡RCD可细分为自噬、铁死亡、细胞焦亡和坏死性凋亡。最近,一些研究揭示了非凋亡RCD与抗肿瘤免疫之间的相互作用,针对自噬、铁死亡、细胞焦亡和坏死性凋亡的靶向治疗结合免疫治疗可能发挥强大的抗肿瘤活性,即使是对ICIs耐药的肿瘤。因此,有必要深入了解抗肿瘤免疫与非凋亡RCD之间的多层次关系,以及非凋亡RCD在提高恶性肿瘤免疫治疗效果方面的潜在靶向应用。
自噬是一种调节机制,可去除不必要或功能失调的细胞成分,并回收代谢底物。针对肿瘤微环境中的应激信号,肿瘤细胞和免疫细胞中的自噬途径发生改变,从而对肿瘤进展、免疫和治疗产生不同影响。
肿瘤微环境(TME),在癌症进展、转移和治疗抵抗中起着关键作用。在TME中,肿瘤细胞中的自噬可由细胞内和细胞外的应激信号诱导,包括代谢应激、缺氧、氧化还原应激和免疫信号。(内容详见 肿瘤免疫和治疗中的自噬)
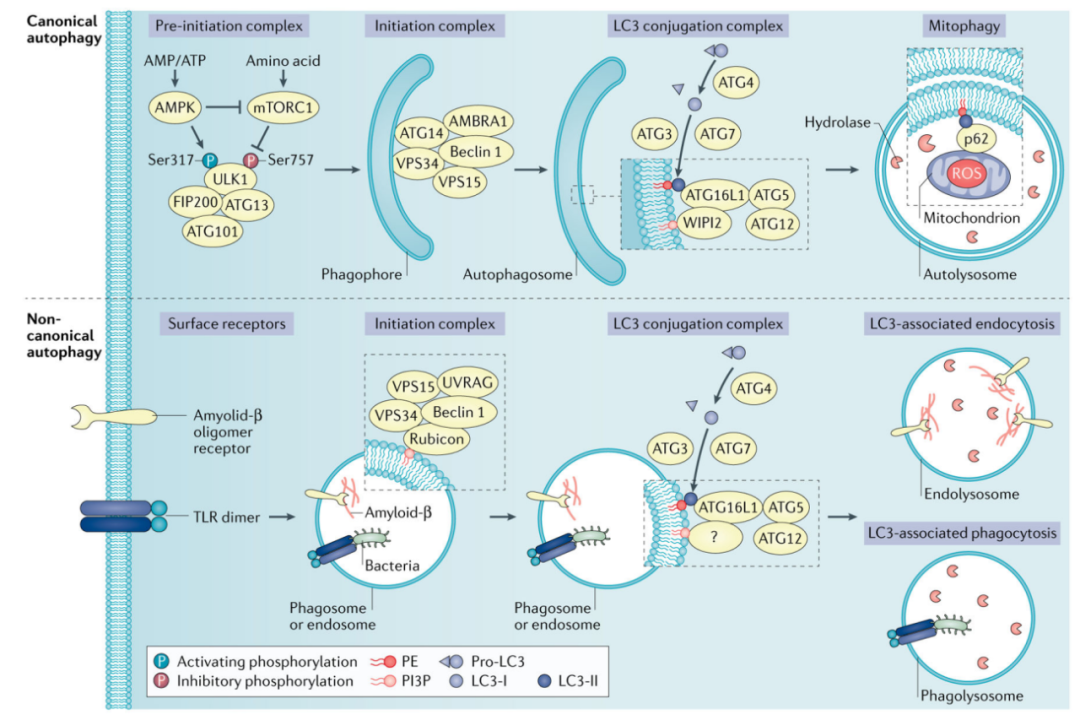

逃避抗肿瘤免疫反应是各种肿瘤的重要生存策略。最近的证据表明,自噬在肿瘤免疫逃避中起着重要作用。有研究发现,胰腺导管腺癌(PDAC)中MHC I类分子的下调是通过选择性自噬降解介导的,抑制自噬释放出强烈的抗肿瘤免疫反应。
另一方面,MDSCs在TME中发挥免疫抑制作用,有研究证明MDSC中的自噬是抑制黑色素瘤抗肿瘤免疫活性的关键机制。MDSC免疫细胞中的自噬是降解MHC II类分子的中心,阻止抗肿瘤T细胞的启动和激活。
此外,自噬作为(癌症)细胞应对威胁性应激源的机制,被认为是癌症治疗中治疗抵抗的重要机制。已有证据表明肿瘤细胞对顺铂的耐药性至少部分是由卵巢癌细胞系中自噬增加介导的。类似的证据表明顺铂、阿霉素和甲氨蝶呤通过抑制骨肉瘤中的自噬来克服化疗耐药性。
自噬抑制剂分为针对ULK1/ULK2或VPS34的早期抑制剂,如SBI-0206965、3MA和wortmannin,以及针对溶酶体的晚期抑制剂,如CQ、羟基氯喹(HCQ)、巴非霉素A1和莫能菌素,CQ和HCQ通过干扰溶酶体酸化抑制自噬体降解。然而,在临床试验中,HCQ单一疗法未能控制晚期胰腺癌患者的肿瘤生长,目前多将自噬抑制与其他癌症治疗相结合,以提高治疗效果。
化疗
癌症中的高自噬通量与化疗反应降低相关,并与癌症患者的低生存率相关。临床前研究表明,抑制自噬可以克服NSCLC、膀胱癌、甲状腺癌和胰腺癌的化疗耐药性。此外,一些研究的结果表明,自噬抑制可能与MEK–ERK信号的抑制产生协同作用。
2014年的一项早期II期研究使用HCQ单一疗法治疗转移性胰腺癌患者,这些患者以前曾通过其他方法治疗,主要终点为两个月的无进展生存期。结果,不同患者的自噬水平不同程度地降低,但主要终点没有显著改善。另一项联合HCQ、吉西他滨和nab紫杉醇治疗晚期或转移性胰腺癌患者的研究也未能证明延长12个月的总生存期。然而,重要的是,采用HCQ的患者显示出了显著且更好的应答率(38.2%对21.1%)。
放疗
自噬在保护肿瘤细胞免受放射治疗引起的细胞死亡方面起着关键作用。在乳腺癌细胞中,辐射诱导自噬相关基因的表达,伴随自噬体的积累。在放疗的同时短期抑制自噬可增强放疗对耐药癌细胞的细胞毒性。同样,低氧通过诱导自噬增强A549肺癌细胞的放射抗性。
在胶质母细胞瘤中,放射治疗通过增加哺乳动物STE20样蛋白激酶4(MST4)的表达诱导自噬,该蛋白激酶通过ATG4B磷酸化来刺激自噬。小分子抑制剂NSC185058(靶向ATG4B)与放疗联合使用,可损害胶质母细胞瘤的颅内异种移植生长,并延长治疗小鼠的生存期。因此,靶向肿瘤自噬可能增强放射治疗的疗效。事实上,在癌症患者的临床试验中,已经利用了自噬抑制剂与放射治疗相结合。
免疫疗法
利用免疫系统是对抗癌症的重要途径。抑制自噬可能会损害系统免疫,因为自噬涉及免疫系统发育和效应T细胞的存活和功能。然而,在黑色素瘤和乳腺癌的临床前模型中,CQ在短时间内对自噬的系统性抑制并没有损害T细胞功能。数据表明,免疫系统可能对某种程度的自噬抑制具有耐受性。然而,鉴于自噬可以调节肿瘤免疫反应,靶向自噬可以提高免疫治疗的疗效,克服免疫治疗的耐药性。
例如,使用抑制剂SB02024或SAR405抑制VPS34激酶活性导致TME中CCL5、CXCL10和IFN-γ水平升高,从而使黑色素瘤和结直肠癌模型中NK细胞和T细胞肿瘤浸润水平升高。在这些模型中,VPS34抑制也逆转了抗PD1或抗PD-L1治疗的耐药性。此外,CQ治疗可阻断自噬介导的MHC I类降解,与双重ICB治疗(抗PD1和抗CTLA4抗体)协同作用,在胰腺癌小鼠模型中产生增强的抗肿瘤免疫反应。
因此,靶向自噬可能增强免疫治疗。目前正在进行HCQ联合免疫治疗治疗不同类型癌症患者的临床试验。此外, 自噬调节可能为使用CAR-T细胞治疗的癌症患者提供一些益处。众所周知,TME是实体瘤中CAR-T细胞浸润和功能的屏障,鉴于自噬抑制可重塑TME并促进TH1型趋化因子的产生,自噬抑制可促进CAR-T细胞向肿瘤的转运,CAR-T细胞的自噬增强可能支持T细胞在TME中的适应性和存活。此外,肿瘤自噬抑制可能导致抗原表达增加,从而增强CAR-T细胞介导的肿瘤杀伤作用。最后,自噬抑制可能改善细胞因子释放综合征并为患者带来临床益处。总的来说,抑制自噬以提高免疫治疗效果的潜力是一个不断探索的有希望的领域。
铁死亡是最近发现的一种程序性细胞死亡,在肿瘤生物学和治疗中发挥着重要作用。这种独特的细胞死亡形式,以铁依赖性脂质过氧化为特征,由脂质、铁和氨基酸代谢等细胞代谢网络进行精确调节。此外,铁死亡也被认为与T细胞介导的抗肿瘤免疫有关,并影响肿瘤免疫治疗的效果。
铁死亡是一种由铁依赖性脂质过氧化引起的调节性细胞死亡。铁死亡的三个关键特征已被破解:膜脂过氧化、细胞内铁的可用性和抗氧化防御的丧失。(内容详见 肿瘤免疫治疗中的铁死亡)
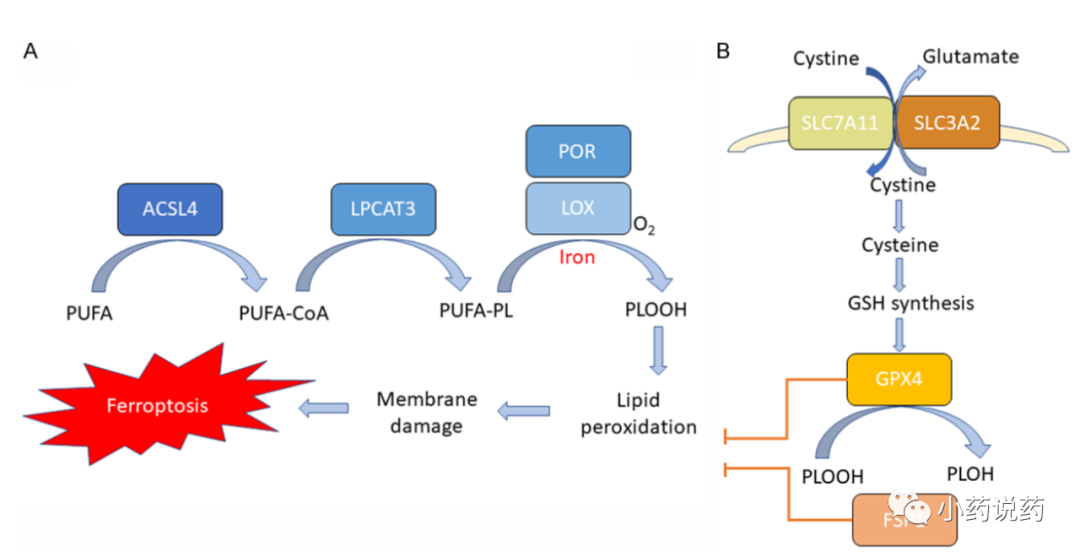
最近发现,铁死亡有助于CD8+T细胞的抗肿瘤作用,并影响抗PD-1/PD-L1免疫治疗的效果。免疫治疗与促进铁死亡的方式相结合,如放射治疗和靶向治疗,可以通过铁死亡产生协同效应,以促进肿瘤控制。
免疫治疗与胱氨酸限制的联合应用
最近有报道称,抗PD-L1免疫疗法激活的CD8+T细胞通过PD-L1阻断后分泌IFN-γ促进肿瘤细胞铁死亡。分泌型IFN-γ显著下调肿瘤细胞中SLC3A2和SLC7A11的表达,导致胱氨酸摄取减少、脂质过氧化增强和随后的铁死亡。胱氨酸/半胱氨酸酶与抗PD-L1协同作用,通过诱导铁死亡可以产生有效的抗肿瘤免疫。
免疫治疗联合靶向治疗
最近的一项研究表明,抗PD-L1治疗的耐药性可通过与TYR03受体酪氨酸激酶(RTK)抑制剂的联合来克服,该抑制剂可促进铁死亡。在抗PD-1耐药的肿瘤中发现TYR03的表达增加。机制上,TYR03信号通路上调关键的铁死亡基因如SLC3A2的表达,从而抑制肿瘤性铁死亡。在TNBC同基因小鼠模型中,抑制TYR03可促进铁死亡并使肿瘤对抗PD-1疗法敏感。该研究揭示了通过使用TYR03抑制剂解除铁死亡是克服免疫治疗耐药性的有效策略。
免疫治疗联合放疗
最近的证据表明,放疗与免疫治疗的协同效应与对铁死亡的敏感性增加有关。辐射已被证明可诱发铁死亡,并且在经辐射处理的癌细胞中观察到铁死亡的遗传和生化特征。其机制涉及辐射诱导的ROS生成和ACSL4的上调,导致脂质合成增强、脂质过氧化增加和随后的膜损伤。因此,放疗的抗肿瘤作用不仅可归因于DNA损伤诱导的细胞死亡,还可归因于铁死亡的诱导。放疗与免疫疗法协同下调SLC7A11,由DNA损伤激活激酶ATM和IFN-γ介导,导致胱氨酸摄取减少、铁死亡增加和肿瘤控制增强。这些研究揭示了铁死亡是免疫治疗和放疗协同作用的一种新机制。
免疫治疗与T细胞铁死亡抑制剂的联合应用
除了诱发肿瘤性铁死亡外,T细胞本身也可能发生铁死亡,这可能会减弱其免疫反应。缺乏GPX4的T细胞迅速积累膜脂质过氧化物,同时发生铁死亡。与癌细胞类似,ACSL4对CD8+T细胞的铁死亡及其免疫功能也是必不可少的。
最近,两项研究表明,CD8+肿瘤浸润淋巴细胞中CD36的表达增加。T细胞固有的CD36促进氧化脂质的摄取并诱导脂质过氧化,从而导致CD8+T细胞功能障碍。这些发现揭示了CD8+T细胞铁死亡是肿瘤免疫抑制的一种新模式,并强调了阻断CD36以增强抗肿瘤免疫的治疗潜力。值得注意的是,该研究还表明GPX4在调节CD8+TIL的抗肿瘤功能中发挥作用。因此,通过GPX4抑制剂在癌细胞中治疗性诱导铁死亡可能会对T细胞产生不必要的靶向效应,并产生不良毒性。
上世纪90年代,在感染鼠伤寒沙门氏菌(S.typhurium)和福氏沙门氏菌的巨噬细胞中首次描述了细胞焦亡。虽然最初被认为是一个凋亡过程,但进一步的研究表明,这种细菌诱导的细胞死亡严重依赖于caspase-1。焦亡细胞与凋亡细胞有一些共同特征,如染色质浓缩和DNA断裂,但可通过其完整的细胞核、孔隙形成、细胞肿胀和渗透溶解来区分。
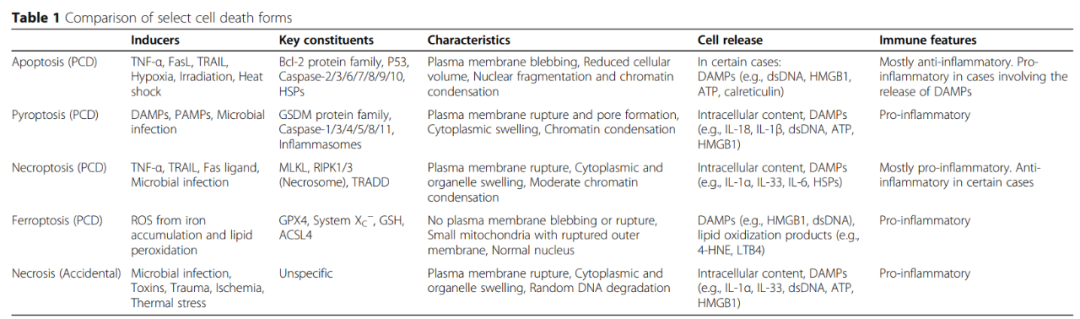
通常,焦亡细胞破裂是通过结合损伤相关分子模式(DAMP)或病原体相关分子模式(PAMP)后,通过半胱氨酸蛋白酶介导的成孔GSDM蛋白活化实现的。这些相同的半胱氨酸蛋白酶也可能直接或间接促进促炎细胞因子的成熟,这些促炎细胞因子与DAMP一起,在释放时启动或维持炎症反应。
尽管已知的细胞焦亡途径的数量在未来可能会增加,但目前已有两条主要途径和几种替代途径被阐明。在主要途径中,细胞焦亡是由GSDMD诱导的,涉及炎症性caspase-1(经典途径)或caspase-4/5(非经典途径)。在替代途径中,最受广泛关注的是GSDME通过caspase-3诱导的细胞焦亡。(内容详见 肿瘤免疫与细胞焦亡)
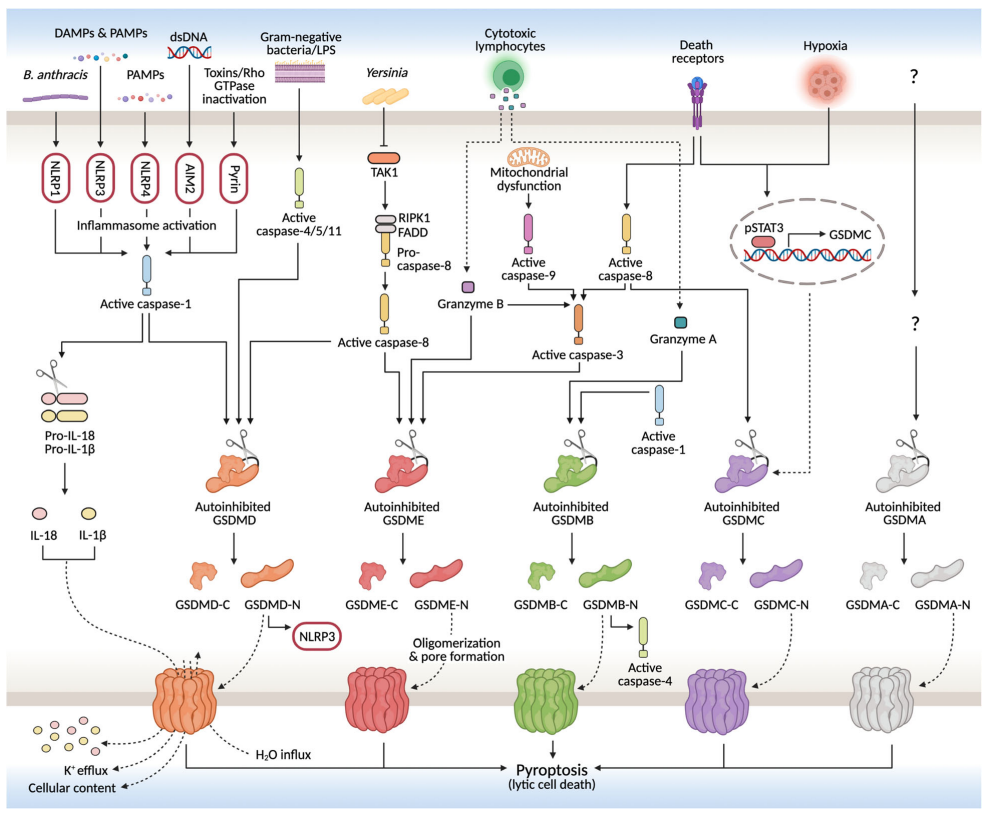
细胞死亡引发适应性免疫反应的能力称为免疫原性细胞死亡(ICD)。与基本上是免疫耐受过程的凋亡不同,细胞焦亡具有诱导强烈炎症反应的分子机制,在某些情况下被认为是ICD的一种形式。虽然细胞焦亡与抗癌免疫之间的联系尚不清楚,但越来越多的研究表明,细胞焦亡介导的肿瘤清除是通过增强免疫激活和功能实现的。
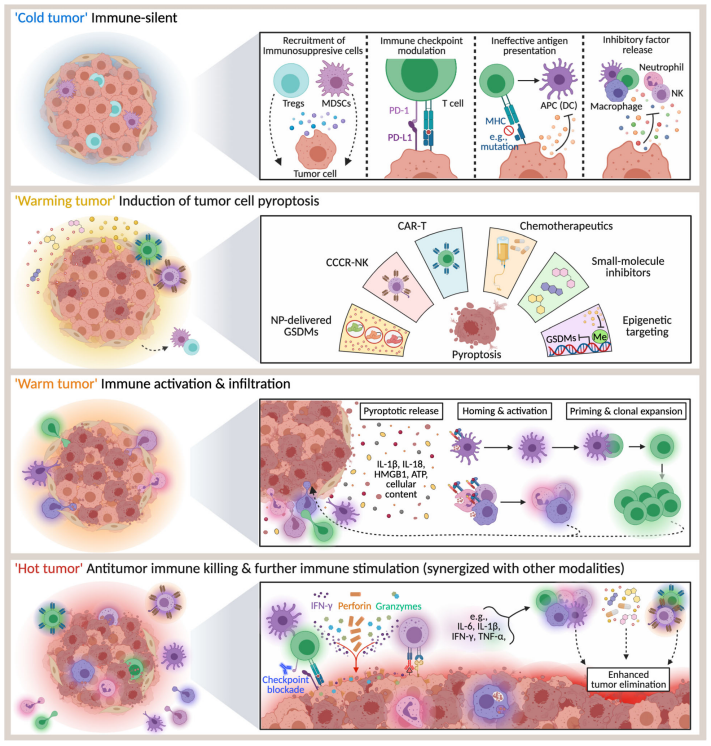
例如,利用肿瘤细胞衍生微粒(TMP)将甲氨蝶呤注入胆管癌(CCA)细胞,以诱导GSDME介导的细胞焦亡,从而激活患者衍生的巨噬细胞,并将中性粒细胞募集到肿瘤部位进行药物导向的肿瘤破坏。此外,当这种甲氨蝶呤TMP输送系统注入肝外CCA患者的胆管管腔时,25%的患者观察到中性粒细胞活化和胆道梗阻的缓解。
此外,还发现GSDME介导的细胞焦亡通过BRAF和MEK抑制剂的组合在黑色素瘤中引起免疫细胞浸润/激活,并导致黑色素瘤消退。在另一种策略中,二甲双胍是治疗2型糖尿病最常用的药物,其可以通过caspase-3间接激活细胞焦亡来抑制癌细胞增殖。
一系列针对KRAS、EGFR或ALK突变型肺癌的小分子抑制剂也被发现,在线粒体固有凋亡途径激活后,通过caspase-3介导的GSDME裂解诱导细胞焦亡。在乳腺癌细胞中,用RIG-1激动剂治疗可触发外源性凋亡途径和细胞焦亡,激活STAT1和NF-κB并上调淋巴细胞募集趋化因子。因此,在小鼠中RIG-1激活后,乳腺癌转移和肿瘤生长的减少伴随着肿瘤淋巴细胞的增加。
几乎所有抗癌免疫治疗策略面临的另一个主要障碍是免疫抑制肿瘤微环境引起的失调。为了解决这个问题,Lu等人设计了含有嵌合共刺激转化受体(CCCR)的NK92细胞,该受体将抑制性PD-1信号转化为激活信号,有效地增强了抗肿瘤活性。在体外,CCCR-NK92细胞通过GSDME介导的细胞焦亡迅速杀死H1299细胞,在体内显著抑制肿瘤生长。
此外,越来越多令人兴奋的研究报告表明,细胞焦亡诱导与PD-1抑制剂协同作用使肿瘤由“冷”变“热”,表明这种组合的巨大潜力。
Deggerev等人于2005年提出的坏死性凋亡是ICD的另一种形式,其中包括FAS和肿瘤坏死因子受体1(TNFR1)等的特定死亡受体(DRs)或toll样受体3(TLR3)等PRR识别来自细胞内外微环境的不利信号,从而引发坏死性凋亡。
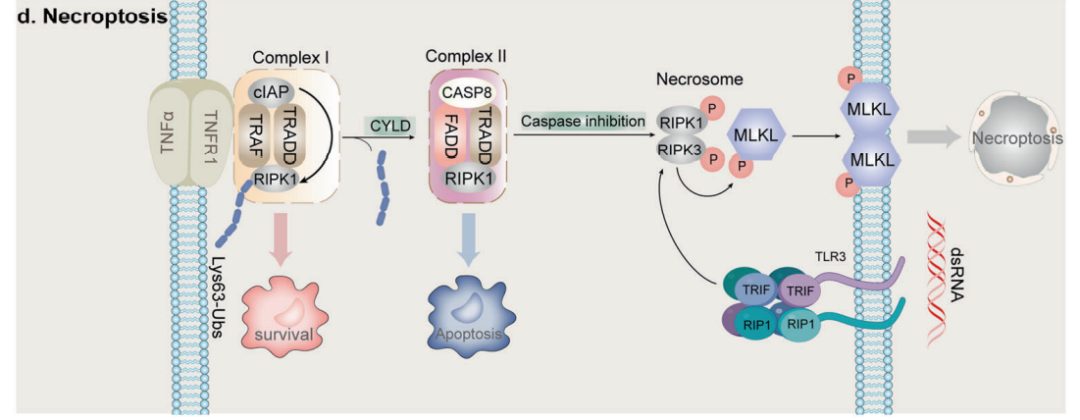
有证据表明,坏死性凋亡在大多数情况下起到肿瘤抑制作用。在一项对60多个癌细胞系的研究中,三分之二的样本显示RIPK3水平降低,这表明癌细胞倾向于逃避坏死性凋亡而存活。此外,坏死性凋亡与癌症预后密切相关,RIPK3的表达是大肠癌患者总生存率和无病生存率的独立预后因素。最近,一项研究表明,RIPK1、RIPK3和MLKL的表达与肝癌患者更好的总体生存率有关。
坏死性凋亡中的效应物如RIPK1和RIPK3可以直接调节免疫细胞的功能,而不依赖于细胞死亡。研究发现,RIPK3介导的磷酸甘油酸变位酶5(PGAM5)激活,通过激活T细胞的核因子(NFAT)的核转位和动力相关蛋白1(Drp1)的去磷酸化,在一个独立于坏死途径的过程中促进自然杀伤T细胞介导的抗肿瘤免疫反应。此外,在同基因黑色素瘤和肺腺癌模型中,将由RIPK3激活的坏死性肿瘤细胞注射到已有的肿瘤中,可以增强抗肿瘤免疫。
坏死性凋亡诱导剂可与ICIs协同抗肿瘤。在黑色素瘤中,SMAC类似物Birinapant使肿瘤细胞对TNF-α介导的T细胞杀伤敏感,并通过调节NF-κB信号通路直接调节免疫细胞功能,包括B细胞、髓源性细胞和细胞毒性淋巴细胞,从而改善对ICIs的反应。类似地,在小鼠肿瘤模型中, SMAC类似物通过RIPK1依赖性细胞死亡激活CD8+T细胞和NK细胞提高了免疫检查点阻断的生存效益。此外,SMAC类似物也被认为可以提高CAR-T细胞治疗急性淋巴细胞白血病的疗效,因为死亡受体信号是CAR-T细胞细胞毒性的关键介质。
非凋亡RCD的研究是一个广泛而迅速发展的领域。一种新的观点是,针对局部肿瘤中的自噬、细胞焦亡、铁死亡和坏死性凋亡,深刻影响TME中浸润的免疫细胞和免疫治疗的反应。非凋亡细胞死亡机制与抗肿瘤免疫之间存在广泛的相互作用。
尽管自噬、细胞焦亡、铁死亡和坏死性凋亡在肿瘤免疫中的作用仍不明确,一些发现提示在不同肿瘤类型和背景下,非凋亡RCD与免疫之间存在更复杂的相互作用,但靶向非凋亡细胞死亡已经越来越成为提高癌症免疫治疗效果的一种有希望的策略。目前,开发更特异的细胞死亡诱导药物,使其作用于肿瘤细胞,对正常组织的副作用最小,这是当务之急。
同时,对这些药物与ICIs以及化疗、放疗和靶向治疗相结合的临床前测试,对于平衡治疗目标和可能的不良反应可能至关重要。未来,应积极鼓励开展联合治疗的临床试验,以评估其疗效和安全性,为后续的深入研究提供更多参考,以使更多的癌症患者受益。
参考文献:
1.Autophagy, ferroptosis, pyroptosis, and
necroptosis in tumor immunotherapy. Signal Transduct Target Ther.2022; 7:
196.
2. Pyroptosis at the forefront of anticancerimmunity. J Exp Clin Cancer Res. 2021; 40: 264.
3. Ferroptosis: a promising target for cancer immunotherapy. Am J Cancer Res. 2021; 11(12): 5856–5863.
4. Autophagy in tumour immunity and therapy. Nat Rev Cancer. 2021 May; 21(5): 281–297.
5. Autophagy in Cancer Therapy-Molecular Mechanisms and Current Clinical Advances. Cancers (Basel). 2021 Nov8;13(21):5575.
编辑:小果果,转载请注明出处:https://www.cells88.com/cells/myxb/18173.html
免责声明:本站所转载文章来源于其他平台,主要目的在于分享行业相关知识,传递当前最新资讯。图片、文章版权均属于原作者所有,如有侵权,请及时告知,我们会在24小时内删除相关信息。
说明:本站所发布的案例均摘录于文献,仅用于科普干细胞与再生医学相关知识,不作为医疗建议。
 微信扫一扫
微信扫一扫  支付宝扫一扫
支付宝扫一扫 