Treg细胞是CD4+T细胞的一个亚群,通过预防自身免疫、控制过度炎症和维持免疫耐受,对免疫稳态至关重要。Treg细胞的免疫调节特性在癌症中会促进肿瘤的进展。对TIL亚群的分析表明,免疫细胞浸润的组成与患者预后之间存在明确的关联。过表达细胞毒性T细胞的TIL通常与良好的预后相关,而TIL中Treg细胞的频率增加与不良预后相关。
Treg细胞的分化和功能依赖于谱系特异性转录因子Foxp3的表达。FOXP3基因功能丧失改变的人类患有免疫失调、多内分泌疾病、肠病、X连锁(IPEX)综合征,表现为广泛的自身免疫和炎症损伤。Treg细胞在胸腺中由一组前体细胞发育而来,这是一个需要T细胞受体(TCR)和IL-2激活的两步过程。
Treg细胞可以通过各种不同的机制抑制免疫反应。CD25是IL-2受体的一种高亲和力亚单位,在Treg细胞上组成性高表达,但在效应T细胞上也上调。Treg细胞上高水平表达的CD25竞争性地结合IL-2,从而剥夺效应T细胞的这种专性生长因子。Treg细胞还表达一系列抑制分子,如CTLA-4,与Treg介导的抑制有关。此外,Treg细胞还可以分泌抑制性细胞因子,如IL-10和TGF-β。
基于这些有效的免疫抑制机制,Treg细胞通常被认为是最有效的抗肿瘤免疫抑制剂。因此,以Treg细胞为靶点的癌症治疗策略也是目前的研究热点。
Tregs可分为两类:天然调节性T细胞(nTregs)和诱导调节性T细胞(iTregs)。两种类型的Tregs都普遍表达 Foxp3。nTregs在胸腺中自然发育,其抑制作用是通过细胞间接触实现的,它们的主要功能是维持正常的免疫耐受和控制炎症反应。
iTregs来源于肿瘤微环境信号诱导的外周原始T细胞,包括肿瘤抗原、细胞因子(如TGF-β)和其他可溶性分子。iTregs通过多种促进肿瘤进展的机制抑制效应T细胞(Teff)、NK细 胞和DC的抗肿瘤免疫作用。
Tregs主要有以下五种功能机制:①Tregs分泌抑制性细胞因子,包括 IL-10、TGF-β 和 IL-35。② Tregs 通过颗粒酶和穿孔素杀死效应细胞。③ Tregs 影响效应细胞的功能:Treg 与效应T细胞竞争消耗 IL-2,从而抑制效应T细胞的生长;Tregs通过产生胞外酶CD39和CD73 促进TME中腺苷的产生,诱效应细胞导的抑制和抗增殖作用;Tregs通过缝隙连接将大量 cAMP 转移到效应T细胞,干扰其代谢。④Tregs通过刺激性和抑制性受体(CTLA-4 或 LAG3)诱导DC耐受,后者通过IDO进一步抑制T细胞的能力。⑤MDSCs和Tregs 产生的因子形成正反馈环,促进增殖,增强抑制环境。
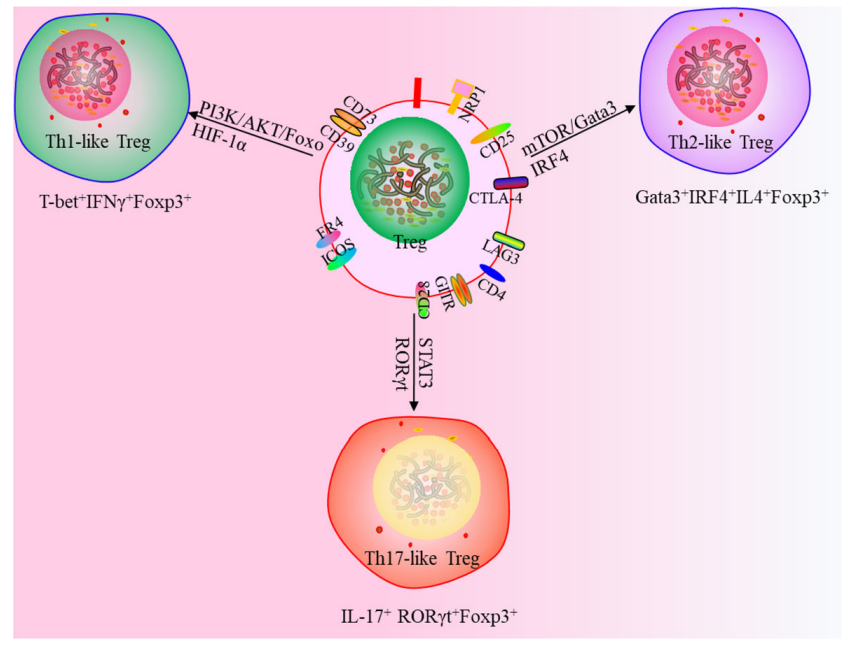
此外,一项研究使用一种新的策略来定义Tregs, Th1样Tregs(T- 234bet+IFNγ+Foxp3+),Th2 样Tregs(Gata3+IRF4+IL4+Foxp3+)和Th17样Tregs(IL-17+RORγt+Foxp3+),这为靶向 Tregs 治疗提供了新思路。
Treg通过多种机制阻碍抗肿瘤免疫反应。在Tregs中,共抑制受体细胞毒性T淋巴细胞抗原4(CTLA-4)通过与APCs中的CD80/B7-1和CD86/B7-2结合干扰共刺激信号,从而抑制效应T细胞中的共刺激受体CD28。
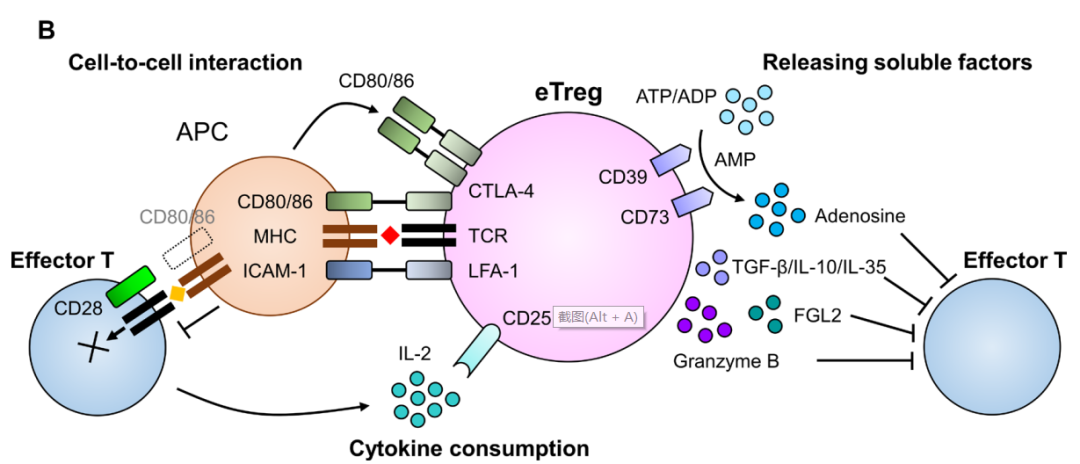
IL-2是Treg和效应T细胞生存所必需的细胞因子。与效应T细胞相比,Treg主要通过由α(CD25)、β(CD122)和γ(CD132)亚单位组成的更高亲和力受体结合IL-2。此外,Tregs比效应T细胞具有更高的CD25表达,而CD25是IL-2的一种高亲和力受体。这使得Treg在利用TME中有限的IL-2方面具有竞争优势。因此,在TME中,Treg的累积量大于效应T细胞。同时,Treg还分泌免疫抑制分子(包括免疫抑制细胞因子)抑制抗肿瘤免疫。Treg产生TGF-β、IL-10和IL-35导致免疫抑制。
表达在Tregs细胞表面的CD39和CD73作为外核苷酸酶,分别将ATP或ADP水解为AMP和AMP水解为腺苷。CD39和CD73产生的腺苷会抑制效应T细胞。当Treg在TME中发生凋亡时,凋亡的Treg通过外核苷酸酶释放大量腺苷,从而产生更强的抗肿瘤免疫抑制。
趋化因子和细胞因子依赖性浸润
Treg具有多种趋化因子受体。趋化因子梯度,如CCR4-CCL17/22,CCR5-CCL5, CCR8-CCL1,和CCR10-CCL28,可参与将Treg招募到TME中。
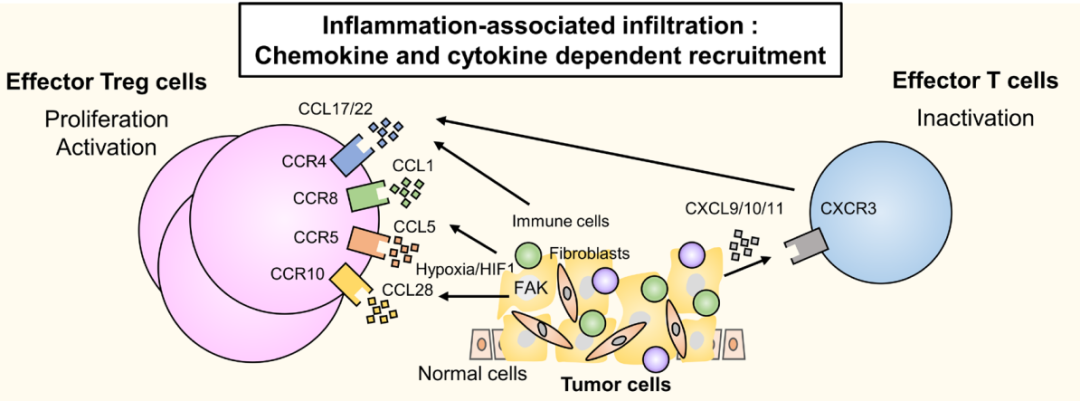
在炎症肿瘤中,炎症细胞产生Treg招募趋化因子,如CCL22。CCR8+Treg被CCR8配体(如CCL1和CCL18)招募到TME等炎症部位,CCL1不仅将CCR8+Treg招募到肿瘤中,还诱导对Treg抑制至关重要的FOXP3、CD39和IL-10的STAT3依赖性上调,从而增强Treg的免疫抑制活性。在一系列细胞因子和趋化因子受体中,与正常组织驻留的Tregs相比,CCR8仅在人类乳腺癌的肿瘤浸润性Tregs中显著上调,表明CCR8是TME中Tregs的一个有希望的治疗靶点,而不会引起系统性自身免疫。
基于TME代谢适应的生存优势
效应T细胞和Treg在正常和炎症条件下使用不同的代谢系统,Treg表现出更强的生存优势。
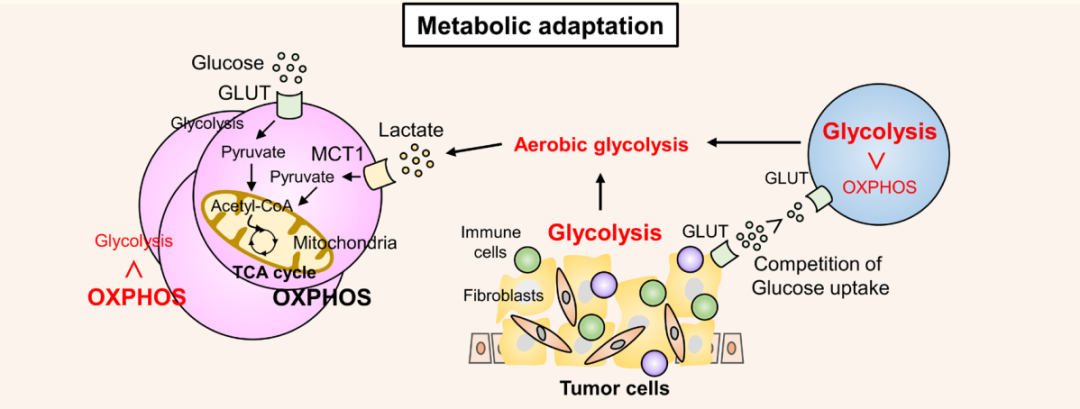
TCR刺激通过磷酸肌醇3-激酶(PI3K)-雷帕霉素(mTOR)信号途径激发特定的代谢程序,导致氨基酸和葡萄糖的吸收增加,从而增强有氧糖酵解。活化的效应T细胞将其代谢程序从氧化磷酸化(OXPHOS)转变为有氧糖酵解,这是其生存和功能所必需的,从而导致效应T细胞和TME中的肿瘤细胞之间的葡萄糖竞争。肿瘤细胞中的代谢重编程将TME转变为营养受限,富含乳酸和低氧环境,不利于效应T细胞的存活和功能。
然而,在TME中,Treg可以在如此恶劣的条件下存活并保持其免疫抑制功能。FOXP3通过抑制糖酵解和促进氧磷和烟酰胺腺嘌呤二核苷酸(NAD+)氧化,因此,Treg可以利用乳酸作为能量来源。当肿瘤浸润性非Treg将丙酮酸转化为乳酸以产生NAD+以维持糖酵解时,TME中的Treg将丙酮酸转化为线粒体中的乙酰辅酶a以触发三羧酸循环,这为Treg提供了比其他T细胞更大的存活益处。
此外,即使在低葡萄糖条件下,Treg还可以利用脂肪酸实现其增殖和免疫抑制功能。研究发现,胃癌中RHOA Y42突变通过上调脂肪酸合成酶(FASN)产生大量脂肪酸,导致TME中更多的Treg和更少的肿瘤浸润性CD8+T细胞。
非炎症肿瘤的浸润机制
虽然炎症肿瘤通常含有Treg,但有时在非炎症肿瘤中也检测到大量Treg,这表明炎症相关浸润以外的机制参与了Treg向TME的募集。
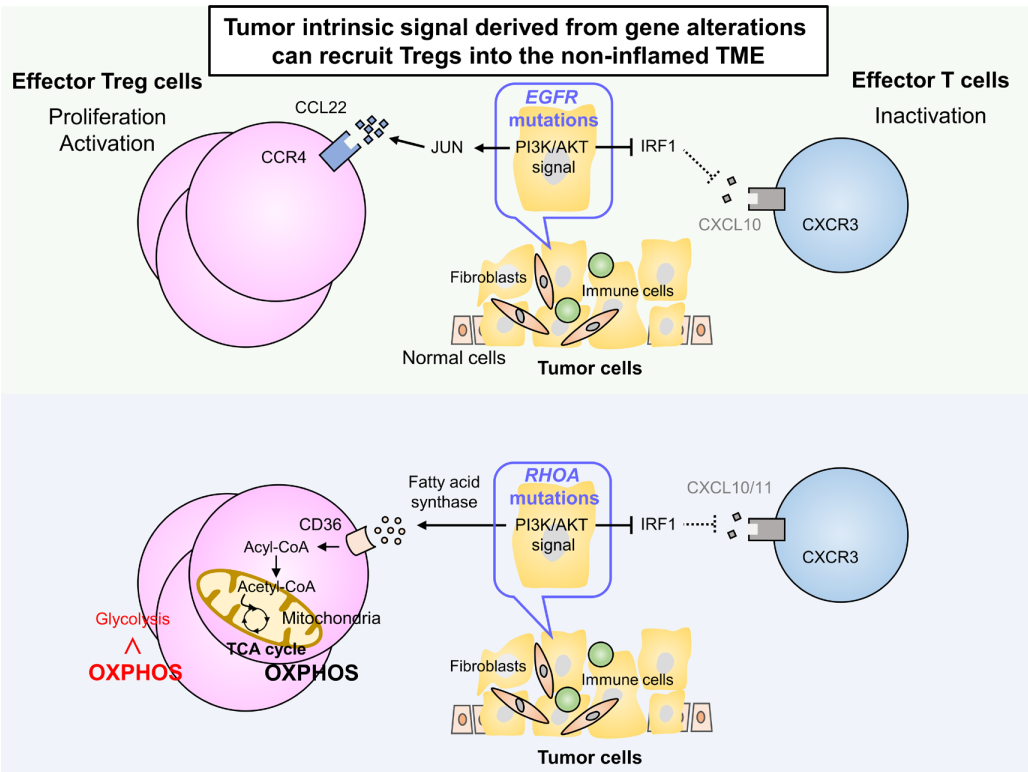
研究发现,某些基因改变可以通过调节下游信号通路来修饰肿瘤细胞产生趋化因子。在肺腺癌中发现的EGFR突变通常与非炎症性肿瘤相关,在这些肿瘤中检测到大量的Treg,而不存在炎症细胞。
EGFR突变通过抑制IFN调节因子1(IRF1)减少CXCL10的产生,这对CXCR3依赖的CD8+T细胞向TME的募集产生负面影响。此外,CCL22的产生通过EGFR下游信号中的JUN诱导而增加,导致CCR4依赖性Treg浸润到TME。
在具有RHOA Y42突变的胃癌中也观察到类似的表型,RHOA Y42突变是一种功能缺失突变,通过抑制IRF1降低CXCL10/11水平。此外,粘着斑激酶的过度激活通过调节趋化因子的产生与Treg浸润和CD8+T细胞排斥相关。
治疗相关的Treg积累
放射治疗和CTLA-4阻断抗体等治疗可能促进肿瘤中Treg的积累,这被认为是一种适应性免疫抵抗机制。由于对辐射的敏感性低于其他淋巴细胞,因此即使在放射治疗后,Treg仍在TME中持续存在,可能导致某些类型癌症发生免疫抑制性TME。在临床前模型中,去除Treg可提高肿瘤对辐射的敏感性并抑制转移。
在小鼠模型中,抗CTLA-4抗体通过抗体依赖性细胞毒性(ADCC)有效地去除Treg,而在患者中的作用机制仍存在争议。免疫组化分析显示,根据治疗前和治疗后活检样本的比较,抗CTLA-4抗体治疗后,Treg数量增加。因此,某些治疗干预可能会增强Treg浸润,从而影响后续治疗的疗效。
新的代谢机制
Tregs活化的核心是支持其生存和功能的脂质代谢的变化,这可由脂肪酸结合蛋白(FABPs)促进。此外,糖酵解和氧化代谢都有助于Tregs的扩增,因为肿瘤内Tregs在葡萄糖摄取方面的相对优势可能促进FA合成。
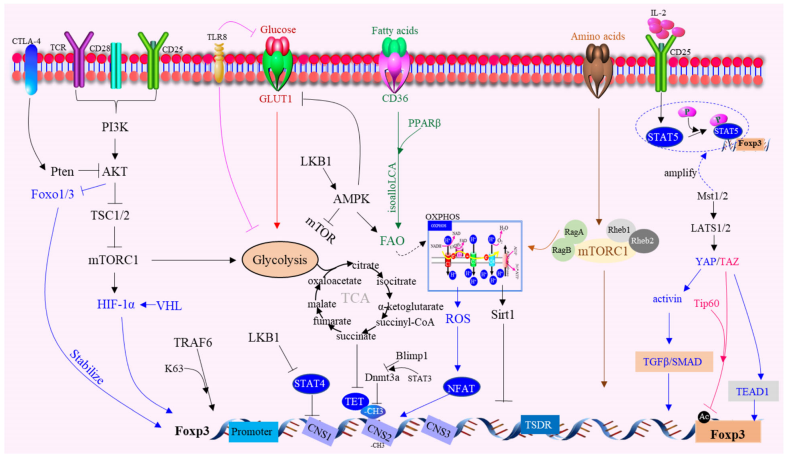
Tregs表现出另外一种独特的代谢特征是线粒体代谢的增加。线粒体转录因子A(Tfam)和线粒体复合体III是维持Tregs免疫抑制功能调节基因表达所必需的。此外,Foxp3缺乏会导致mTORC2信号转导的失调,并增强有氧糖酵解和氧化磷酸化。
值得注意的是,CD36在肿瘤内的Tregs中被选择性上调,作为一种中枢代谢调节剂。CD36通过PPARβ信号精细调节线粒体适应性,重编程Tregs以适应富含乳酸的TME。在抗PD-1治疗过程中,CD36靶向诱导了额外的抗肿瘤反应。
肝激酶B1(LKB1)调节Tregs的代谢和功能适应度,并作为DCs免疫原性的关键抑制剂发挥作用。在Tregs中,LKB1的特异性缺失可导致一种以Th2型显性反应过多为特征的致命性炎症性疾病,不仅破坏Tregs的生存、线粒体适应度和代谢,还可诱导免疫调节分子的异常表达,包括PD-1和TNF受体超家族蛋白的GITR和OX40。
新的遗传机制
Helios可以增强人类胎儿CD4+幼稚T细胞向Tregs的优先分化,作为在炎症反应期间稳定Tregs的关键转录因子。肿瘤中Helios缺陷的Tregs获得效应T细胞功能,并通过上调对自身抗原具有高度亲和力的效应细胞因子来促进对癌症的免疫反应,如GITR/PD-1表达增加和对自身抗原的反应性增强。
Foxp1是一种forkhead转录因子,Foxp1的缺失会导致Tregs的功能受损,TSDR、CNS2、CNS3均为Foxp3相关的作用元件,调控着Foxp3的表达和功能。
YAP是Hippo通路的辅活化因子,在Tregs中高度表达,并促进Foxp3的表达和功能。TAZ是一种诱导Hippo信号转导的TEAD转录因子的辅激活因子,它通过减少组蛋白乙酰转移酶Tip60介导的Foxp3的乙酰化而减弱Tregs的发育。
核受体Nr4a是维持Treg基因程序的关键转录因子,参与Treg介导的TME抗肿瘤免疫抑制。促自噬蛋白AMBRA1也是T细胞的一个关键调节因子。此外,SUMO特异性蛋白酶3(SENP3)介导的BACH2去SUMO化阻止BACH2的核输出,从而抑制与CD4+T效应细胞分化相关的基因,并稳定Treg特异性基因特征。
新的分子机制
在Tregs的分子机制方面也取得了一些新的进展。Tregs具有丰富的IL-2受体(IL-2R)表达,依赖于活化T细胞产生的IL-2,提示Tregs对IL-2的消耗与其抑制功能有关。并且IL-2R依赖的转录因子STAT5的激活在Tregs的抑制功能中起着重要作用。
丝氨酸-苏氨酸激酶Mst1被鉴定为Tregs中IL-2-STAT5活性的信号依赖性放大器,Tregs中Mst1和Mst2的高活性对于防止肿瘤抵抗和自身免疫是至关重要的。
在T细胞活化过程中,Tregs中Foxp3的磷酸化可以通过TAK1 Nemo样激酶(NLK)信号通路进行调节。另外,Tregs激酶TAK1的特异性缺失可以减少外周淋巴器官中Tregs的数量,而E3泛素化连接酶TRAF6缺乏的Tregs在体内功能紊乱。
ST2被认为是IL-33的唯一受体,表达ST2的Tregs对IL-33有反应,并且在IL-33刺激下Tregs的百分比增加,特别是Foxp3+GATA3+Tregs。在结直肠癌(CRC)的小鼠模型中,肿瘤浸润的Tregs优先上调ST2,IL-33/ST2信号与肿瘤负荷呈正相关。
激活的Tregs表面表达糖蛋白-A为主的重复序列(GARP),它结合并激活潜伏的TGF-β。此外,整合素α4β1的激活也可增加Tregs的抑制能力。
Tregs稳态通过Tregs内在和外在机制与黏膜相关淋巴组织1(Malt1)功能密切相关。相比之下,CARD11-BCL10-MALT1(CBM)信号对于以Malt1蛋白酶依赖的方式介导Tregs的抑制功能至关重要。
抗CTLA-4单克隆抗体,如ipilimumab,通过Treg靶向改善TME的免疫特征。然而,在临床研究中的详细作用机制仍然存在争议。在小鼠模型中,抗CTLA-4单克隆抗体诱导的抗肿瘤免疫反应完全依赖于通过Fc介导的ADCC对Treg的消耗。而在人类中,Treg的消耗与伊普利单抗抗肿瘤的主要疗效无关。此外,靶向肿瘤内Tregs高表达分子的双特异性抗体,如CTLA-4和OX-40或CTLA-4和GITR,它们可能通过有效地消耗体内的Treg来提高抗肿瘤疗效。
由于肿瘤内Treg比其他效应T细胞表达更高水平的CD25,CD25可能是Treg耗竭的关键靶点。一些针对CD25的治疗策略已经被FDA批准。例如,IL-2与白喉毒素的融合蛋白denileukin diftitox用于皮肤T细胞淋巴瘤。denileukin diftitox可与表达CD25的细胞结合,并通过白喉毒素的细胞毒性作用杀死它们。
新的抗CD25抗体RG6292被开发用于选择性地清除Tregs,而不会干扰效应T细胞中的IL-2信号,目前正在进行临床评估(NCT04158583)。靶向CD25的近红外光免疫治疗(NIR)也可能是肿瘤局部Treg去除的一种有希望的方法。此外,还开发了ADCT-301,一种抗CD25的抗体偶联药物,用于靶向表达CD25的淋巴瘤。它业可作为另一种在肿瘤中去除Tregs的选择,目前正在进行临床试验(NCT03621982)。
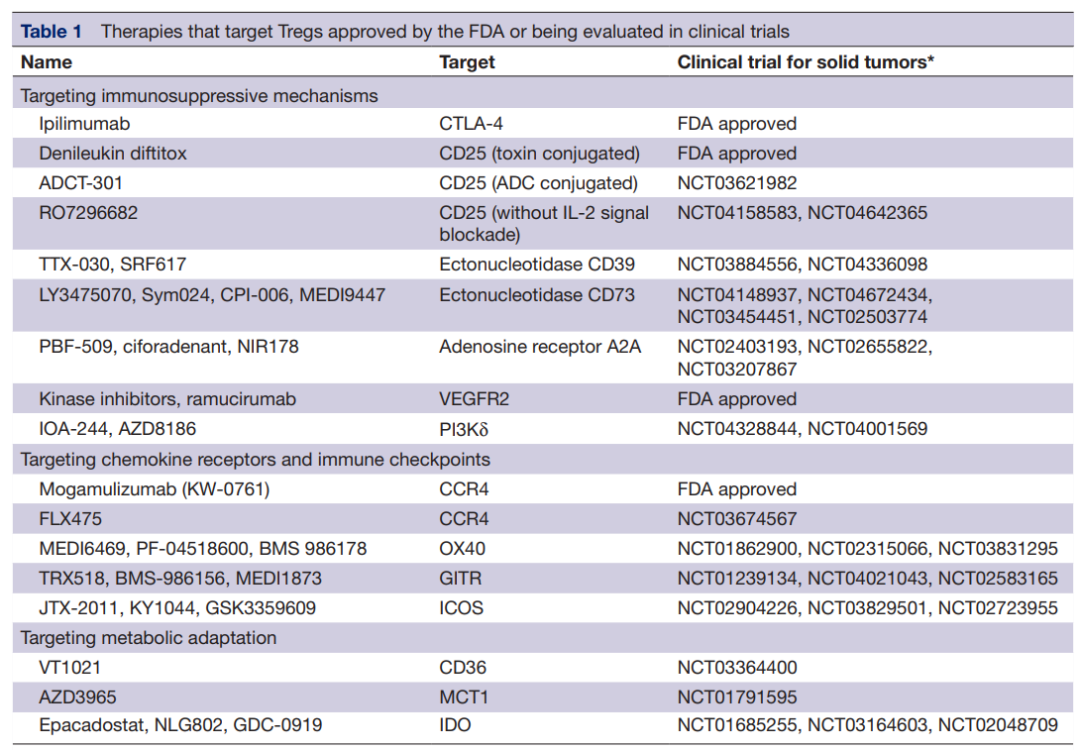
通过Tregs表达的外核苷酸酶CD39和CD73靶向腺苷途径也可能是增强抗肿瘤免疫的一个有希望的靶点。阻断外核苷酸酶活性的抗CD39和抗CD73抗体,如TTX-30、MEDI9447和BMS-986179,目前正在进行临床试验(NCT03884556、NCT03742102和NCT02754141)。除了抑制腺苷的产生外,腺苷受体A2AR的小分子抑制剂也可防止腺苷依赖性T细胞抑制。这些抑制剂的临床试验正在进行中(NCT02655822、NCT04089553和NCT02403193)。
此外,靶向VEGFA-VEGFR2轴可能通过减少TME中的Treg增殖和浸润来激活抗肿瘤反应。已经证明,抗VEGFR2抗体ramucirumab可以减少胃癌患者eTreg的增殖。以FOXP3为靶点的疗法也有开发。AZD8701是FOXP3的反义寡核苷酸,在体外实验和人源化小鼠模型中部分降低了FOXP3及其下游转录分子的表达。AZD8701的临床试验正在进行中(NCT04504669)。
抗CCR4抗体mogamulizumab可减少实体瘤患者中CCR4+Treg的数量。在一项临床试验中,mogamulizumab联合抗PD-1抗体(nivolumab)治疗的患者中证实了Treg的缺失,这表明该组合是癌症免疫治疗组合中一个有希望的选择。
由Treg高度表达的免疫检查点分子,如OX40、GITR和ICOS,也可能成为治疗靶点。一些研究表明,这些受体的刺激降低了Treg的免疫抑制功能,导致效应T细胞的激活。作为OX40激动剂的抗体,如MEDI6469134(NCT02274155),GITR,如MK-4166135(NCT02132754),以及ICOS,如KY1044136(NCT03829501),目前正在开展临床研究。
在炎症和非炎症肿瘤中,TME中Treg浸润、激活和存活的多种机制都已被揭示,影响TME中Treg浸润、激活和存活的机制可能因患者而异。肿瘤细胞中的基因改变不仅决定免疫原性和炎症状态,而且也有助于调节TME中的免疫细胞浸润和生存,例如基于趋化因子的Treg浸润和基于代谢变化的Treg激活,这些都可以被特异性激酶抑制剂靶向。因此,TME中主要导致Treg浸润的机制需要通过基于基因改变的免疫学和代谢谱来表征,并以精准免疫治疗为目标。基于机制的Treg靶向治疗有望改善目前的免疫治疗。
参考文献:
1.Mechanisms of regulatory T cellinfiltration in tumors: implications for innovative immune precision therapies.J Immunother Cancer. 2021; 9(7): e002591.
2. Regulatory T cells in tumor microenvironment: new mechanisms, potential
therapeutic strategies and future prospects. Mol Cancer. 2020; 19: 116.
3. Regulatory T Cells in Tumor Microenvironment and Approach for Anticancer Immunotherapy. Immune Netw. 2020 Feb; 20(1): e4.
编辑:小果果,转载请注明出处:https://www.cells88.com/cells/myxb/18036.html
免责声明:本站所转载文章来源于其他平台,主要目的在于分享行业相关知识,传递当前最新资讯。图片、文章版权均属于原作者所有,如有侵权,请及时告知,我们会在24小时内删除相关信息。
说明:本站所发布的案例均摘录于文献,仅用于科普干细胞与再生医学相关知识,不作为医疗建议。
 微信扫一扫
微信扫一扫  支付宝扫一扫
支付宝扫一扫