

由于同一个细胞因子可以由不同的细胞产生,同一个细胞在不同的条件下又能产生不同的细胞因子,同时再叠加上细胞因子可以参与自分泌信号、旁分泌信号和内分泌信号。因而我自己看的时候常常被各种细胞因子给搞晕。刚好趁着这次机会,将零散的资料汇总了一下,希望起个抛砖引玉的作用。
细胞因子包括趋化因子、干扰素、白细胞介素、淋巴因子和肿瘤坏死因子。常见的细胞因子见下
肿瘤免疫中的相关细胞因子
肿瘤发生
长期以来,IL-1被认为与炎症诱发的癌症发生有关。pro-IL-1β通过病原体识别受体(如Toll样受体、C型凝集素受体或RIG-I样受体)对危险相关分子模式(DAMPs)和病原体相关分子模式(PAMPs)作出快速反应。
在慢性炎症的背景下,IL-1α和IL-1β 可能直接促进致癌介质(如NO和ROS)的产生, 并且促进IL-6,IL-11以及IL-22的生成和释放。IL-6、IL-11与IL-22一起快速诱导STAT3的磷酸化,STAT3信号的激活可在多种类型的癌症中观察到,诱导增殖、存活、上皮-间质转化(EMT)以及转化细胞的迁移。
IL-1β 与TGF-β 一起诱导T辅助细胞17(TH17)分化,在经IL-23刺激后分泌IL-17A和IL-17F(IL-17A/F)。IL-17通常介导伤口愈合信号,也可能加剧新生肿瘤的生长。
IL-33可以创造一个自我扩增的致瘤生态,促进新生肿瘤的发展。如鳞状细胞癌模型所示,一旦细胞发生转化,它们就会获得致瘤能力。肿瘤起始细胞可分泌IL-33,进而导致肿瘤相关巨噬细胞浸润,并促进TGF-β产生致瘤信号。
肿瘤生长与进展
恶性肿瘤具有一些重要特征,即持续增殖、炎症、血管生成、主动侵袭和迁移,而这也是伤口愈合的标志,因此,肿瘤可能会恶意利用旨在组织修复得细胞因子信号。
IL-1不仅促进炎症诱导的癌变,而且有助于肿瘤的侵袭性和血管生成。
一些癌症类型被证明过度表达某些细胞因子,例如IL-6或IL-11,这些细胞因子可能以自分泌的方式激活PI3K–AKT–mTOR通路,从而上调糖酵解,诱导代谢重编程;同时激活NF-κB、RAS-RAF-MAPK和STAT3信号通路,这些通路会促进EMT、增殖、迁移、凋亡减少以及IL-8, VEGF等细胞因子的产生,从而诱导血管生成。
其他细胞因子,如IL-1β, IL-13、IL-17、IL-22、IL-23和IL-35也可诱导EMT,从而导致肿瘤进展。
肿瘤分泌的IL-8可以诱导多形核白细胞(PMN)的募集,与单核细胞一起,它们分化成髓源性抑制细胞(MDSCs),抑制T辅助细胞1(TH1)的反应。
TGF-β 与IL-33一起促进Treg细胞的分化, Treg细胞会抑制抗肿瘤反应。另外,TGF-β与IL-6可以促进TH17细胞分化产生IL-17,进一步促进MDSC的募集和分化。
肿瘤免疫监视
固有免疫
抗原提呈细胞(如DC和MΦ)可以产生IL-12和IL-15,从而促进NK细胞和CTLs的细胞毒活性,诱导IFN-γ释放。
IL-18通过其在NK细胞中高表达的受体发挥作用并触发IFN-γ产生、细胞毒性和FAS配体(FASL)表达。
IL-28A、IL-28B和IL-29是与IL-10家族有较远亲缘关系的细胞因子,同时也是干扰素,因此也被称为“III型干扰素”或“λ-干扰素”。它们通常介导固有免疫抗病毒活性,但也直接诱导恶性细胞凋亡。
适应性免疫
T细胞反应的增殖、存活、分化和终结主要由IL-2、IL-7和IL-15控制,而IL-3是淋巴细胞祖细胞存活和增殖所必需的。
CTL和效应TH1细胞是抗肿瘤适应性免疫的主要介质。来源于DC细胞的IL-12提供了驱动T-bet表达的必要信号,从而促进效应TH1细胞和CTLs的分化。与NK细胞中的信号传导类似,IL-2、IL-15和IL-18与IL-12协同触发IFN-γ 的产生以及CTL和TH1细胞的直接细胞毒性。
此外,DCs与M1巨噬细胞一起产生维持TH1细胞极化和IFN-γ扩增所必需的IL-12。M1巨噬细胞分泌的IL-10、巨噬细胞和DC细胞分泌的IL-27以及H17细胞和滤泡辅助T细胞产生的IL-21也可能增强细胞毒性。
肿瘤免疫逃逸
尽管肿瘤免疫监视和免疫编辑具有强大的作用,但恶性细胞仍可能进化以逃避抗肿瘤反应,并利用外源性免疫抑制机制促进肿瘤进展。
Treg
高亲和力IL-2信号与TGFβ 诱导Foxp3,FOXP3可促进Treg细胞分化和IL-10的产生,从而建立免疫抑制的TME。IL-33也可以直接促进TGF-β诱导Treg细胞分化,抑制IFN-γ并促进肿瘤中Treg细胞的稳定性。
Treg细胞在TME中的调节作用主要由IL-10、IL-35和TGF-β的分泌介导。IL-10具有抗炎作用,而IL-35诱导抑制性表面受体(包括PD1、LAG3、TIM3、TIGIT和CD244)的表达,并限制T细胞记忆的形成。
TH17型反应与髓系抑制物
IL-1β刺激TH17细胞和γδ T细胞产生IL-17,其招募大量免疫抑制性粒细胞。此外,在肺癌中,IL-23将第1组固有淋巴细胞转化为产生IL-17的ILC3s,从而促进IL-17介导的肿瘤细胞增殖。
此外,肿瘤衍生的IL-8(也称为CXCL8)为乳腺癌患者的髓样细胞提供趋化信号,并对免疫治疗产生耐药性。同样,炎症小体激活后产生的IL-18驱动多发性骨髓瘤中MDSCs的生成,从而产生免疫抑制机制。
TH2型反应
肿瘤和间质细胞可能分泌促进TH2细胞和M2巨噬细胞极化的细胞因子,从而抑制抗肿瘤TH1细胞极化和应答。同样,IL-33诱导的第2组固有淋巴细胞(ILC2)在小鼠黑色素瘤模型中被证明对抗NK细胞功能。反过来,乳腺癌癌相关成纤维细胞分泌的IL-33是ILC2和TH2型应答的有效增强剂,可能诱导TCR非依赖性分泌IL-13并招募免疫抑制性粒细胞。
免疫细胞代谢重编程
癌细胞可诱导TME免疫细胞的代谢重编程和代谢系统的改变,从而诱导炎症反应向免疫抑制反应的转变。依赖NF-κB诱导激酶(NIK)的效应T细胞糖化开关在小鼠黑色素瘤模型中被证明是抗肿瘤免疫的关键。来源乳酸的糖酵解在TME中的积累可增加氧化磷酸化,并与IL-4一起诱导抗炎再编程。
此外,TME中乳酸的积累以及氨基酸和葡萄糖的消耗与TGF-β信号结合可诱导Treg细胞极化,增强免疫抑制和T细胞衰竭,从而使肿瘤对免疫治疗产生抵抗。胰腺导管腺癌中,基质细胞释放IL-6,增加肿瘤细胞的糖酵解和乳酸外溢,导致巨噬细胞M2极化,抗PD1治疗的疗效降低。
因此,肿瘤源性物质的积累和慢性炎症反应使抗肿瘤免疫无法抑制肿瘤的生长和进展,导致肿瘤生长失控和远处转移,限制了靶向治疗的效果。
自免中的相关细胞因子
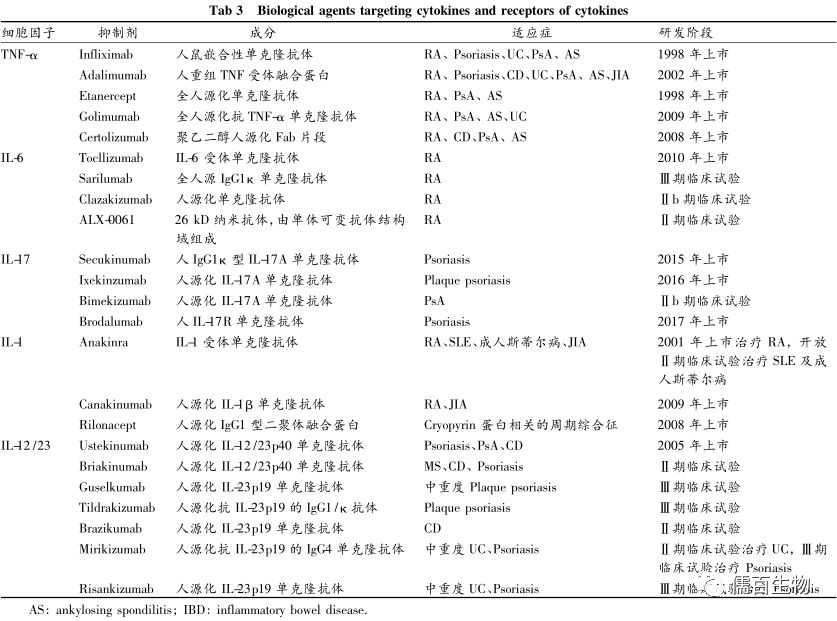

TNF-α
TNF-α在RA滑膜中表达的增加和转基因小鼠关节炎的发生提示TNF-α在慢性炎症中的致病作用,它是一种依赖于TNF-α受体(TNFR)结合的多效细胞因子,TNFR1几乎在所有有核细胞上构成型表达,主要负责TNF-α的炎症功能,而TNFR2仅在特定的细胞类型上表达,如骨髓源性抑制细胞、Treg细胞和单核细胞,与TNF的调节功能相关。目前可用的抗TNF-α药物同时抑制TNFR1和TNFR2。
由于TNF-α的调节作用,TNF-α阻断可能会矛盾地诱导Th1/Th17细胞扩增和IFN反应失调,这可以解释抗TNF治疗过程中的治疗失败、自身抗体产生和反常的银屑病。因此,研究人员正在研究更具有选择性的治疗方法来抑制TNFR1和增强TNFR2。
IL-6
IL-6最初被认为是由T细胞分泌的“B细胞分化因子”或“B细胞刺激因子”。首个靶向IL–6R的tocilizumab在RA中显示出显著的临床益处,甚至优于adalimumab的疗效,目前被批准用于治疗RA, JIA,AOSD、巨细胞动脉炎和基于其调节全身过度炎症的能力的CRS。最近,抗IL-6治疗的研究已拓展到SLE、视神经脊髓炎和系统性硬化症中的应用。
IL-6信号转导通过IL-6R和糖蛋白130 (gp130)的复合体完成。gp130的二聚作用可依次激活多条信号通路,这些信号通路可移位到细胞核,并控制与细胞生长、增殖和炎症相关的基因转录。为了干扰IL-6信号通路,抗IL-6治疗可靶向IL-6、IL-6R、gp130、JAK、STAT3。基于对副作用的考虑,抗IL-6单抗、抗IL-6R单抗和JAK抑制剂已被用于阻断IL-6信号转导,而使用可溶性gp130抑制IL-6/IL-6R复合物的潜力正在研究中。
IL-17
IL-17A,俗称IL-17,是CD4+ T细胞Th17亚群的标志细胞因子。在生理状态下,Th17细胞通过招募中性粒细胞、宿主防御细菌和真菌、皮肤和黏膜部位的组织修复等方式参与先天免疫。在病理条件下,IL-17A促进自身免疫。结合IL-17A的是一种独特的胞内胞质结构域,称为IL-17RA和IL-17RC,招募接头蛋白Act1,从而触发TNF-受体相关因子(TRAF) 6的泛素化,以及随后转录因子激活。越来越多来自动物和人类研究的证据表明,IL-23/IL-17轴是多种自身免疫性疾病的绝佳治疗靶点。
目前的抗IL -17疗法包括IL-17A的单抗(secukinumab和ixekizumab)和IL-17RA (brodalumab)。抗IL -17治疗在银屑病、PsA和As中产生了显著的反应。出乎意料的是,这种方法在Th17依赖的RA患者中并没有显示出显著的临床疗效。这种结果可以解释为疾病本身的异质性,或IL-17A在RA分期中的不同致病作用。
IL-23/IL23A & IL12B
IL-23是一种由DCs和活化的巨噬细胞分泌的促炎细胞因子。作为IL-12家族的一员,IL-23是由IL-12共有的p40亚基(IL-12/IL-23p40)和IL-23独有的p19亚基(IL-23p19)组成的异源二聚体。虽然IL-12和IL-23共享一个结构亚基,IL-23在自身免疫性疾病的发病机制中更为关键和广泛。最重要的是,IL-23通过维持Th17标志基因、抑制抑制因子、上调IL-23R表达、诱导效应基因等途径稳定Th17细胞的致病特征。它还可作为自分泌因子促进DCs和巨噬细胞的促炎功能。
阻断IL-23信号通路可以通过靶向抑制IL-12/IL-23p40或IL-23p19来实现。作为一种抗IL-12/IL-23p40单抗,ustekinumab可阻断IL-12和IL-23,而抗IL-23p19单抗(guselkumab, tILdrakizumab, risankizumab和mirikizumab)仅抑制IL-23。
与抗IL-17治疗相似,抑制IL-23对治疗银屑病和PsA有效。在克罗恩病中,ustekinumab成功改善了临床结果,抗IL-23p19单抗的临床试验正在进行中。
I型IFN
I型IFN包括IFN-α、IFN-β、IFN-ε、IFN-κ和IFN-ω,I型IFN与IFN-α/β受体(IFNAR)结合,IFNAR由IFNAR1和IFNAR2组成。研究表明,IFN-α在自身免疫性疾病特别是SLE的发病机制中起着重要作用,在SLE中,IFN-α通过刺激髓系DCs、Th1细胞和B细胞以及抑制Tregs来促进促炎症反应。
CAR-T中用到的细胞因子
虽然活化的CAR-T细胞产生细胞因子,如IL-2,但反复暴露于肿瘤细胞后产生减少,而一些对T细胞效应功能重要的细胞因子,如IL-12和IL-15,要么在极低水平上,或者根本不是由T细胞产生的。
由于这些限制,研究人员开始优化设计,将细胞因子工程入CAR,使得CAR-T可以分泌需要的细胞因子,来增强CAR-T细胞激活信号3,目前已经有十多种细胞因子或者细胞因子受体用于CAR的设计。
IL-2和IL-15
L-2和IL-15促进免疫细胞增殖和抗凋亡蛋白的转录,但对这两种细胞因子的反应在自然杀手(NK)细胞和T细胞亚群之间是不同的。
高亲和力的IL-2R在调节性T细胞(Tregs)和激活的CD4和CD8T细胞表达,尽管记忆性CD8T细胞和NK细胞对IL-2的反应,但程度小一些。相反,记忆性CD8T细胞和NK细胞对IL-15反应最强,而Treg和初始CD4+和CD8+T细胞则不是。
因为使用IL-2会扩增调节性T细胞,抑制抗肿瘤活性,因而逐渐减少使用。而且长时间使用IL-2会产生AICD效应及诱导T细胞分化为终末的细胞毒性T细胞,对T细胞的持久性不利。目前IL-2主要还是作为药物,但用或者和CAR-T联合使用。
IL-15除了作为药物单独使用,也可以将IL-15基因工程入CAR,提高了CAR-T细胞的抗肿瘤活性,目前在CD19、GPC-3、CLL-1、GD2和IL-13Ra2 CAR-T临床前研究显示具有抗肿瘤活性。
IL-7
IL-7皮下给药,可诱导循环CD4+和CD8+T细胞数量的剂量依赖性增加,而没有IL-2和IL-15治疗引起严重的不良事件,如毛细血管渗漏综合征。
除了促进T细胞的扩增,IL-7还被证明通过优先扩张初始T细胞,增加TCR库的多样性,进一步增强抗肿瘤活性。
将IL-7和CCL9基因工程入CAR,也开始进入多项临床试验。
IL-9
CAR-T细胞在体内耗竭标志物表达上调,持久性差,是其治疗失败的因素之一。因而降低或阻断耗竭标志物(主要是免疫检查点),是延长CAR-T体内存活时间,增强抗肿瘤活性的策略之一。
IL-9被发现可以增加CAR-T中央记忆表型,降低耗竭标志物(PD-1,LAG-3等),增加持久性。
休斯顿卫理公会研究所的科学家,尝试将CAR-T诱导为分泌IL-9的T9表型,发现其在小鼠模型中存活时间和抗肿瘤活性都大大增强(文献3)。
将IL-9基因工程入CAR,理论上也是一个可行的方案,不过现在还没有看到相关报道。
IL-12
IL-12促进T细胞和NK细胞增殖、细胞毒性,以及分泌高水平的IFNγ、TNF-a和GM-CSF。IL-12通过直接诱导IFNγ,上调T-bet和IL-12Rb2的表达,促进Th1表型分化。
IL-12已被用作一种单药进行免疫治疗,临床使用主要问题为限制性毒性。除了单药使用,也与CAR-T联合使用。
目前有基因工程IL-12 CAR-T,进入临床。CAR-T分泌IL-12增强T细胞核NK细胞功能,促进IFN-γ释放,促进巨噬细胞和DC细胞功能。
IL-18
IL-18由巨噬细胞、树突状细胞、上皮细胞和其他类型的细胞产生,与异二聚体受体(IL-18Ra和IL-18Rb)相互作用,IL-18R在NK细胞和有抗原经验的T细胞上表达。
对于肿瘤,IL-18是一种双功能分子。通过MyD88和NF-kB发出信号,具有致瘤作用,如促进血管生成、肿瘤转移和增殖,但由于其在Th1反应,则很大程度上具有抗肿瘤活性。
与IL-12或抗原刺激相结合,IL-18诱导Th1和CD8+T细胞产生IFN-γ和细胞毒性效应分子,激活NK细胞和巨噬细胞等。
L-36-γ
IL-36a、IL-36β和IL-36γ是IL-1超家族的成员,在临床前模型中显示出强大的抗肿瘤活性。
将IL-36-γ基因工程入CAR,可以显著的改善增殖和持久性。表达IL-36γ的CAR-T细胞能够增强肿瘤小鼠脾巨噬细胞和树突状细胞上MHCII类和CD86的表达,提示IL-36在抗原提呈细胞的成熟中发挥作用。在小鼠模型,显示出超强的抗肿瘤活性(文献7)
IL-21
和其他γ链细胞因子不同,IL-21偏向激活STAT3而非STAT5,同时也通过PI3K和MAPK通路介导增殖。
小鼠模型研究,和表达IL-2, IL-7,或 IL-15 CAR-T细胞相比,表达IL-21的CAR-T细胞对总体生存率的改善最好。尽管这些细胞表达的Bcl-2、IFN-γ和TNF-a水平较低,但可以维持较低分化的效应记忆T细胞(TEM)亚群和/或增强长期持久性。
NCT04715191,Interleukin-15 and -21 Armored Glypican-3-specific Chimeric Antigen Receptor Expressed in T Cells for Pediatric Solid Tumors,研究单位:Baylor College of Medicine
IL-23
IL-23是一种双亚基细胞因子,可促进记忆T细胞和Th17细胞的增殖。
北卡罗来纳大学教堂山分校Xingcong Ma等的工作表明,IL-23改善实体肿瘤中CAR-T细胞功能,且比IL-15的副作用低。CAR/TCR信号通路上调IL-23R和IL-23p19的表达,但不上调IL-23-IL-12p40亚基的表达。将T细胞转导,使其组成性表达IL-12p40,导致激活诱导IL-23的表达,而不是IL-12,并促进T细胞增殖。
小鼠模型中,显示良好的抗肿瘤活性(文献6)。
IL-24
IL-24主要在免疫细胞中表达,在乳腺癌、肺癌、淋巴瘤等多种肿瘤中作为一种有效且几乎普遍存在的癌症抑制因子。此外,肿瘤中直接注射表达IL-24的腺病毒(Ad.mda-7;INGN-241)后,晚期癌症患者中具有抗癌活性和安全性。
中南大学发表在Signal Transduction and Targeted Therapy上的结果显示,IL-24 CAR-T在小鼠模型中,具有强的抗肿瘤活性。
细胞因子对于CAR-T活性、增殖、持久性等非常重要。除了经典的IL-2、IL-12、IL-15等已经作过很多临床尝试的细胞因子外,现在IL-7、IL-18、IL-21等显示出促进CAR-T活性、持久性或者毒副作用少等优势,也开始进入临床试验。IL-24、IL-36等细胞因子也开始在临床前的模型中进行尝试,显示出良好的抗肿瘤活性。
编辑:小果果,转载请注明出处:https://www.cells88.com/cells/myxb/16542.html
免责声明:本站所转载文章来源于其他平台,主要目的在于分享行业相关知识,传递当前最新资讯。图片、文章版权均属于原作者所有,如有侵权,请及时告知,我们会在24小时内删除相关信息。
说明:本站所发布的案例均摘录于文献,仅用于科普干细胞与再生医学相关知识,不作为医疗建议。
 微信扫一扫
微信扫一扫  支付宝扫一扫
支付宝扫一扫 