

介绍
免疫或免疫突触是淋巴细胞通过细胞间相互作用与抗原呈递细胞(APC)、抗原特异性靶向细胞和其他淋巴细胞进行通信的中心作用机制。
在正常的T细胞生物学中,小(直径约5µm)循环的幼稚CD8+ T细胞找到抗原呈递细胞(APC),并通过T细胞表面上聚集的TCR与新或APC表面上的非自身肽抗原负载MHC分子。
这种相互作用导致CD8+ T细胞在接下来的4-5天内分化和活化成“武装”的抗原特异性杀伤T细胞,这些细胞装载了充满溶细胞蛋白、颗粒酶和穿孔素的颗粒 。这些引发的抗原特异性T细胞扩增和增殖,直径增加到约10µm,诱导更复杂的细胞骨架系统“加载”细胞膜附近的溶细胞颗粒并表达额外的激活和反应受体。在定位表达非自身或新抗原的细胞(通常是肿瘤细胞或感染细胞)后,T细胞与靶细胞形成溶细胞突触并释放其溶细胞毒素以杀死这些细胞 。此外,FasL等T细胞膜溶解因子也可以在突触中起作用以诱导靶细胞凋亡。
免疫突触,也称为超分子激活簇(SMAC),负责启动和完成APC和T细胞之间的细胞间反应。SMAC形成三个同心环,类似于“靶心”(图 4),中央SMAC (cSMAC) 形成中心环,由外围SMAC(pSMAC) 和远端SMAC(dSMAC)环绕。每个环都有自己特殊的功能和结构。cSMAC含有一定浓度的TCR和共刺激分子CD28,负责伴随突触形成的关键T细胞激活信号事件,pSMAC含有一系列粘附分子,如LFA-1,可稳定细胞间相互作用和dSMAC 由丝状肌动蛋白组成,有助于在突触上施加机械力。
免疫突触有多种形式,每一种都有自己的特殊功能。经典的免疫突触,例如与 APC 相互作用的幼稚CD4+ T细胞,是一种抗原识别突触。CD8+细胞和NK细胞可以形成刺激性突触,导致细胞因子分泌或抑制性突触。CD8+ T细胞和NK细胞也可以与靶细胞形成溶细胞突触,从而导致杀伤,这是T细胞和NK细胞重定向疗法的基础。
溶细胞突触与经典的免疫突触非常相似,但具有额外的活动来驱动靶细胞杀伤。这些包括肌动蛋白和微管引导的裂解蛋白定位、指导细胞毒性蛋白(如穿孔素和颗粒酶)分泌的信号以及使用dSMAC的机械力来增强穿孔素活性并将细胞毒性杀伤集中在一个定向的、极化的方式。
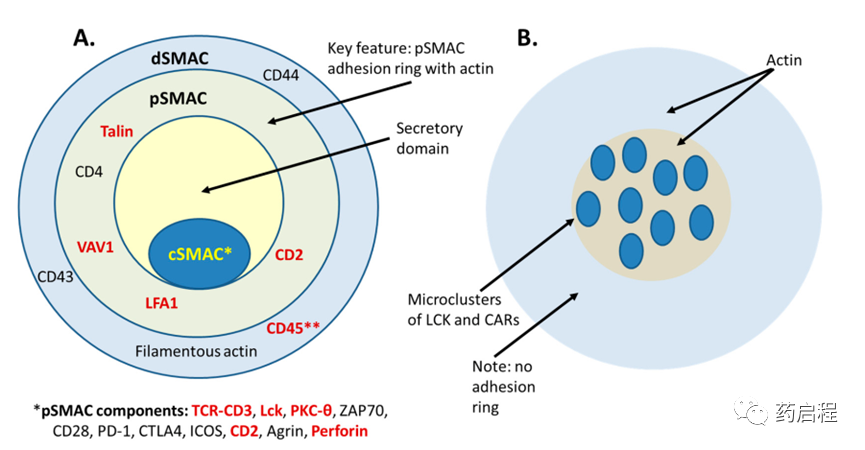

正常TCR-pMHC突触与CAR-T和TRBA诱导的突触
一段时间以来,与TCR复合物-MHC相互作用信号传导到T细胞相关的精心策划的事件一直是一个研究领域。TCR复合物是TCR α/β或δ/γ异二聚体的复杂结构,类似于抗体,可精确识别肽-MHC (pMHC)复合物。然而,与这种相互作用相关的细胞内信号来自该复合体的其他成员,即CD3ε、δ、γ和ζ。这些链通过跨膜和铰链/茎结构域之间形成的离子键与细胞表面的TCR α/β(δ/γ) 异二聚体特异性结合(图 5)。
虽然以前认为只是一个集群驱动的事件,它通过ITAM内关键残基的磷酸化来驱动下游信号传导,但现在人们认识到,这种相互作用期间的重要结构变化会驱动下游事件的强度和持续时间。此外,在TCR α/β复合物 (TCR δ/γ复合物不需要 CD4/CD8 共受体参与来发挥作用)。CD4和CD8的细胞内结构域也与Src激酶LCK结合以提供额外的信号传导,其功能尚不完全清楚。总的来说,这种复杂的结构已经进化到控制哺乳动物和其他生物体内最复杂的细胞活动之一。TRBA和CAR的目标一直是尽可能地模仿这种复杂性。
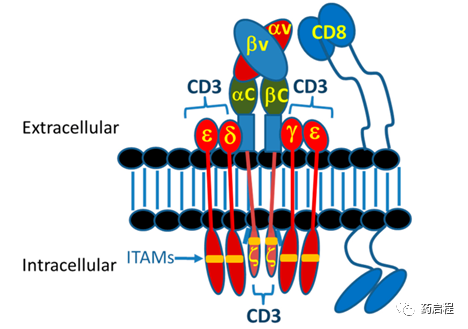
图 5. T 细胞受体 (TCR) 复合物示意图。正常的TCR-pMHC相互作用在1–100 µM范围内,聚集TCR的固有亲和力提供了所需的吸引力[11]。MHC对呈递肽的亲和力对天然 T 细胞杀死和根除肿瘤的能力具有深远的影响。当在体外测定中发现肽-MHC亲和力小于10 nM时,表明识别这些pMHC复合物的T细胞能够引起肿瘤排斥[116]。另一方面,当肽-MHC 亲和力>100 nM 时,肿瘤复发,表明 T 细胞不能杀死这些肿瘤细胞[116]。CAR-T细胞和TRBA均独立于该参数发挥作用。
T细胞/靶细胞突触由亲和力和受体-靶点密度之间的微妙平衡驱动,涉及自然 TCR-pMHC 相互作用、TRBA 和 CAR。这些相互作用的结果可能受到天然调节受体的影响,例如 CD45 异构体,它可以自然地下调从 TCR 或 CAR 发出的信号 [117]。嵌入细胞膜中的 CD45 是一种复杂的、高度差异剪接的、具有不同细胞外大小的分子,可以干扰这些基于突触的相互作用并阻止下游信号传导 [118]。低亲和力的相互作用,典型的 TCR-pMHC 相互作用,很容易受到 CD45 异构体的影响,作为一种天然的安全机制来防止不希望的 T 细胞活化 [117]。然而,TCR 和 pMHC 之间更高的亲和力相互作用或多重相互作用可以克服这些影响 [117]。在设计 TRBA 和 CAR 时也必须考虑类似的因素。
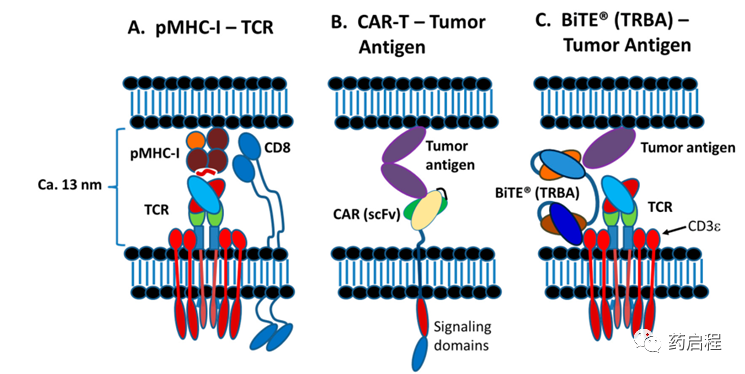
在突触形成过程中,T细胞和APC之间的间距已被测量为5-25nm,T细胞 TCR 和负载肽的MHC (pMHC)复合物之间形成的突触的正常间距已显示约为13纳米(图 6)。实验上强制延长细胞膜之间的距离会降低TCR的激活和响应 ,这对于TRBA和CAR-T来说都是一个关键问题,如下所述。
对于天然 CTL,已表明只需 1-3个肽-MHC/TCR相互作用即可触发溶细胞杀伤事件。然而,在这些情况下,复杂的SMAC复合体既不是必需的,也不是完全形成的。此外,最近用NK细胞(另一种溶细胞性淋巴细胞)证明,NK细胞系仅产生约200 个穿孔素阳性颗粒,而溶细胞突触处的一次脱颗粒事件导致仅释放约20个颗粒,仅约 2-4其中实际上是杀死目标细胞所必需的。因此,基于细胞溶解突触的杀伤机制非常有效且对激活敏感。
TRBA诱导的突触或CAR-T细胞与靶细胞之间形成的突触与天然突触相比如何?Baeuerle及其同事的一项研究表明,由抗EpCAM×CD3ε BiTE® TRBA聚集在一起的T细胞和 EpCAM+ 细胞之间形成的突触在其结构和功能上与正常的溶细胞性T细胞突触高度相似,包括形成同心环和许多相同蛋白质标记的存在,例如LCK、PKC-θ、LFA-1、VAV1、Talin、CD3、穿孔素和CD2 (图 4)。此外,还单独证明了 TRBA 诱导的突触还具有经典的突触标志,包括靶点聚集、ZAP70易位和从 cSMAC 中排除负调节蛋白CD45。
虽然由TRBA的功能形成的突触似乎与正常MHC/TCR介导的突触高度相似,但 CAR-T突触似乎与正常T细胞突触显着不同。CAR-T突触不像SMAC那样高度组织化,而是杂乱无章、不完整的信号簇,缺乏明确的结构。此外,CAR-T突触不需要LFA-1稳定,也不会形成特征性 pSMAC。因此,很明显,CAR-T细胞与其靶细胞形成的免疫突触在结构上与经典免疫突触和TRBA形成的突触不同(图 4)。CAR-T突触和经典pMHC/TCR突触之间的这些结构差异也会导致功能差异。与经典突触相比,CAR-T细胞产生更快的近端信号传导并更快地将溶酶体募集到免疫突触,这表明它们能够产生比TCR介导的杀伤更快速的杀伤反应。
此外,与TCR驱动的T细胞相互作用相比,它们具有明显更快的解离速率,即突触的溶解和从靶细胞脱离的速度。在突触形成后TCR介导的杀伤与CAR-T杀伤的时间过程比较中,CAR-T信号强度比TCR信号强度更大且上升更快,穿孔素和颗粒酶释放更快(开始后两分钟内的峰值释放CAR-T与TCR的3分钟)和分离明显更快(CAR-T 为 5 分钟,TCR 为 7 分钟)。因此,CAR-T似乎比CTL更快地杀死靶细胞,然后更快地继续前进。此外,在体外已经证明,CAR-T细胞可以在最初识别靶抗原后约 25 分钟内杀死靶抗原阳性肿瘤细胞。
最近,熊等人证明CAR-T突触的强度(通过肌动蛋白和裂解颗粒的定量测量)比 4 小时杀伤试验的任何一种细胞因子产生更能预测杀伤效果,这表明CAR-T突触的重要性是CAR-T功能,即使它的结构与传统的免疫突触显着不同。
如上所述,溶细胞性TCR/pMHC 突触、TRBA诱导的突触和CAR-T靶细胞突触都会导致靶细胞死亡,通常是通过穿孔素和颗粒酶诱导的细胞凋亡,尽管对于 CAR-T 细胞, FAS/FAS-L 轴也参与其中。然而,正如结构差异所预期的那样(图 6),与TCR-pMHC突触相比,由 TRBA和CAR-T形成的不依赖TCR的突触具有一些独特的特征。下面将重点介绍其中的一些差异。
首先,TRBA和CAR-T细胞均独立于TCR/pMHC发挥作用,因此不需要在靶细胞上表达MHC受体。这在一项研究中得到证实,其中BiTE®s可以杀死EpCAM阳性、MHC I类阴性细胞系,表明 BiTE®s 可以在完全没有 MHC T细胞识别分子的情况下杀死靶细胞。其次,TCR对pMHC的亲和力通常在1-100 µM 的范围内,而CAR或TRBA对其目标的亲和力通常低于100 nM,通常低于10 nM。
第三,由于通过 TCR/pMHC相互作用而导致的T细胞活化部分受pMHC在靶细胞上的表达控制,这些靶细胞通常以非常低的拷贝数存在。另一方面,TRBA和CAR-T细胞的靶点通常在1000秒到10,000秒之间,有时甚至更高。然而,即使靶细胞上的抗原密度非常低,例如只有200个靶分子/细胞,TRBA仍能引发T细胞杀伤。因此,即使在低目标密度下,至少在某些情况下,TRBA仍然可以是有效的杀伤剂。
当抗原密度达到低至 200 个靶分子/细胞的水平时,CAR-T 细胞似乎也能够杀死靶细胞。在直接比较 TRBA 和类似 CAR-T 细胞活性的研究中,CAR-T细胞似乎比类似的 BiTE® 更有效地杀死低抗原密度的靶细胞。有趣的是,与TCR信号传导发现的分层阈值相似,触发 CAR-T 杀伤所需的抗原密度(100-200个靶点/细胞)明显低于触发 CAR-T 细胞因子释放所需的抗原密度(~ 5000个目标/细胞)。因此,似乎与 TCR 反应类似,CAR-T细胞激活杀伤、增殖和细胞因子释放存在不同的阈值,完全反应需要至少 5000个抗原/细胞的密度。
另一方面,与TRBA或pMHC/TCR介导的突触形成有很大不同,CAR-T 细胞可能直接导致靶细胞抗原丢失,从而导致抗原密度低。在最近的一项研究中,证明 CAR-T 细胞通过 trogocytosis 机制降低靶细胞上的抗原密度,这是将靶向抗原转移到 CAR-T的过程。这一过程不仅降低了靶细胞上的抗原密度,而且一旦CAR-T细胞获得靶抗原,它们本身就可能成为CAR-T相残的靶点,可能导致缺乏持久性 [130]。
第四,据计算,对于具有fMIC50的最有效的BiTE®,需要“两位数”(即
最后,通过TCR-pMHC相互作用的T细胞激活通过CD8的募集得到增强,并结合了“信号 2”共刺激途径。相反,T细胞受到检查点相互作用的负调控,例如 PD-1/PD-L1 和 CTLA4/CD80-86 。TRBAs和 CAR-T细胞参与的T细胞都可以独立于信号 2发挥作用。然而,共同刺激分子如 CD28 和 CD137(4-1- BB)。对于CAR-T细胞,CAR本身设计有来自 CD28、OX40、ICOS 和/或 4-1BB 的细胞内共刺激信号结构域,在 CAR 与靶细胞结合时提供共刺激信号 [94,125,134]。
与 CTL的调节类似,TRBA和 CAR-T细胞参与的 T 细胞可以受到检查点途径的抑制,例如 PD-1/PD-L1 和 CTLA4/CD80-86。考虑到这一点,目前正在进行临床试验,将B细胞淋巴瘤或白血病的治疗与抗 CD19 BiTE®、Blincyto®、抗 PD-1或抗PD-1和抗CTLA4相结合抗体。
同样,抗PD-1和/或抗CTLA4检查点抑制剂联合疗法也正在与CAR-T疗法一起进行临床测试,以努力减轻CAR-T细胞的检查点抑制。在某些情况下,第四代CAR-T 细胞正在被设计为表达抗体或抗体片段,这些抗体或抗体片段可以以自分泌/旁分泌方式发挥作用,以阻断检查点抑制剂如PD-1或PD-L1或工程化到CAR-否定PD-1/PD-L1抑制途径的T细胞显性阴性PD-1。在这两种情况下,PD-1的抑制作用都被阻断,增加了CAR-T细胞的效应功能和持久性。此外,通过敲除其内源性PD-1基因改造的CAR-T细胞的临床试验正在进行中,以测试其假设的改善功效。
如上所述,虽然在CTL(TCR/pMHC)、TRBA或CAR-T细胞范例中激活 T 细胞的机制存在显着差异,但也有许多相似之处。这些包括通过重定向的 T 细胞与靶细胞形成突触、作为连环杀手发挥作用的能力、它们的杀伤机制(例如,定向释放溶细胞蛋白如穿孔素和颗粒酶以杀死靶细胞)、它们通过共刺激的调节和检查点抑制途径及其增殖和分泌细胞因子的能力。
Bispecific T-Cell Redirection versus Chimeric Antigen
Receptor (CAR)-T Cells as Approaches to Kill Cancer Cells
Antibodies 2019, 8, 41; doi:10.3390/antib8030041
编辑:小果果,转载请注明出处:https://www.cells88.com/cells/myxb/10179.html
免责声明:本站所转载文章来源于其他平台,主要目的在于分享行业相关知识,传递当前最新资讯。图片、文章版权均属于原作者所有,如有侵权,请及时告知,我们会在24小时内删除相关信息。
说明:本站所发布的案例均摘录于文献,仅用于科普干细胞与再生医学相关知识,不作为医疗建议。
 微信扫一扫
微信扫一扫  支付宝扫一扫
支付宝扫一扫 