

介绍
尽管嵌合抗原受体(CAR)T细胞在治疗血液系统恶性肿瘤方面已显示出显着成果(1),但针对实体癌的有效CAR T治疗方法的开发仍然是一项挑战,很大程度上是由于难以确定最佳目标表面抗原。极少的抗原是真正的肿瘤特异性抗原,与正常组织的靶点/肿瘤外交叉反应会导致致命的毒性(2-5)。此外,即使鉴定出高度肿瘤特异性的抗原,这些靶标也常常是异质表达的,选择性CAR靶向可以使抗原阴性的肿瘤细胞逃逸(6)。因此,通常需要新颖的肿瘤识别策略,其可以应对特异性和异质性的双重挑战,以扩大用于安全有效地攻击实体癌的治疗窗口。
在胶质母细胞瘤(GBM)中发现了这种双重挑战的具体例子(图1A)。表皮生长因子(EGFRvIII)是一种高GBM特异性新抗原,存在于一部分GBM患者中(7-10)。但是在先前的临床研究中,以EGFRvIII CAR靶向GBM导致了肿瘤复发,尽管有效杀死了EGFRvIII+细胞,但EGFRvIII表达的高度异质性却使EGFRvIII-肿瘤细胞得以逃脱(6、11、12)。相反,替代的神经胶质瘤相关表面抗原,包括Aphrin A型受体2(EphA2)和IL13受体α2(IL13Rα2),在绝大多数GBM细胞中表达(13-15),但特异性不完善。尽管它们在正常脑组织中不表达,但在一些非肿瘤,非脑组织中却以低水平表达(13-15)。总而言之,找到单一的,既特异性又均质的理想表面GBM抗原具有挑战性。
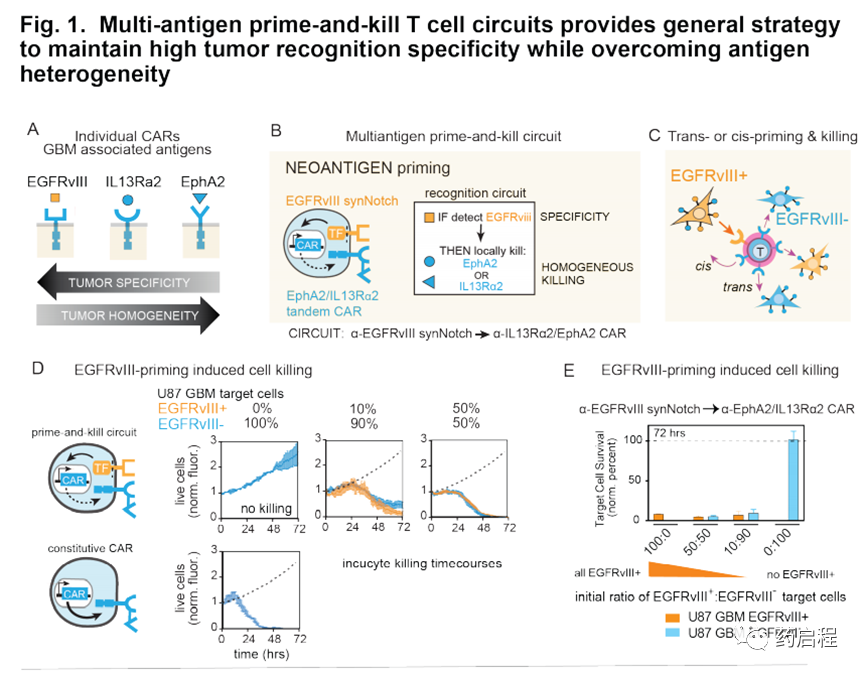

互补的抗原识别回路
识别多种抗原组合的T细胞为抗原异质性和特异性问题提供了可能的解决方案。我们先前开发了“启动和杀死”回路,其中synNotch受体(一种工程受体,当其识别其同源抗原时会激活转录输出(16),诱导针对杀伤抗原的CAR的表达(17、18)。在这里我们假设,通过仔细选择引发和杀死抗原(分别是synNotch和CAR配体),这样的回路可能导致杂交识别行为,这可能提供一种在抗原特异性和异质性之间进行权衡的方法。
我们采用两种不同的策略-用肿瘤特异性但异质抗原(例如EGFRvIII)引发(图1B-E)或用脑特异性抗原(例如髓鞘少突胶质糖蛋白(MOG))引发(图1F-H )。这些抗原引发的启动子随后被用于局部诱导识别更均一的抗原EphA2和IL13Rα2的CAR的表达。EphA2和IL13Rα2的肿瘤特异性不完善,使其成为常规单靶CAR T细胞治疗方法的非理想靶标。
但是,如果通过启动抗原提供更高的肿瘤选择性,这些抗原可以作为有效的杀伤靶标。我们假设,用引发和杀死回路工程化的T细胞可以诱导局部CAR驱动的细胞毒性,仅局限于引发细胞附近,从而避免了在表达杀伤抗原但缺乏引发抗原的远距离正常组织中的随意杀伤。这种类型的回路在空间上整合了对两个不完美但互补的抗原靶标的识别:启动抗原提供特异性,而杀伤抗原则确保治疗攻击的均一性。在所有这些回路中,为了实现均匀杀伤并进一步降低肿瘤逃逸的可能性,我们利用了同时靶向两种杀伤抗原EphA2或IL13Rα2的串联CAR(13、19)。串联CAR用作OR门,其胞外区域包含一个α-EphA2单链抗体和一个IL13突变蛋白(IL13配体的变体,与IL13Rα1相比与IL13α2的亲和力更高)(19、20)。
为了克服这种异质性,一个关键问题是由一个细胞(EGFRvIII+)引发的致敏和杀伤性T细胞是否可以杀死另一个相邻的靶细胞(EGFRvIII-)–我们称其为反引发过程/杀死(图1C)。
为了进行转杀的首次测试,我们使用了经过工程设计以稳定表达启动抗原EGFRvIII的U87“启动”GBM细胞和天然U87“靶标”细胞(内源性表达EphA2和IL13Rα2杀伤抗原,但对EGFRvIII阴性)(11,12,21,22)。我们以不同的比例混合了U87-EGFRvIII+和U87-EGFRvIII-细胞,以概括在GBM患者(10-100%的启动细胞)中观察到的不同水平的异质性,然后测试了启动细胞的存在是否诱导了靶细胞的杀伤(图1D,E)。
我们发现,用α-EGFRvIIIsynNotchàαIL13Rα2/ EphA2 CAR引发和杀死回路改造的CD8+ T细胞可以在体外有效杀死EGFRvIII-靶细胞,即使低至10%EGFRvIII+引发细胞也是如此(图1D,E)。相反,在不存在引发细胞的情况下,未观察到对EGFRvIII-靶细胞的杀伤。在这些分析中,我们追踪了72小时内杀死两种不同肿瘤细胞群体(EGFRvIII-和EGFRvIII+)的动力学。用低至10%的启动细胞观察到有效杀伤,尽管与用50%的启动细胞观察到的杀伤相比稍慢(p=.0149;t检验)。我们还验证了用模型抗原进行反式杀伤的有效性,从而显示了反式启动/杀伤的耐用性。所有这些体外杀伤研究均表明,有效地进行了启动引发/杀死,并且EGFRvIII引发的回路因此代表了一种防止因异质性导致的肿瘤逃逸的有前途的策略。
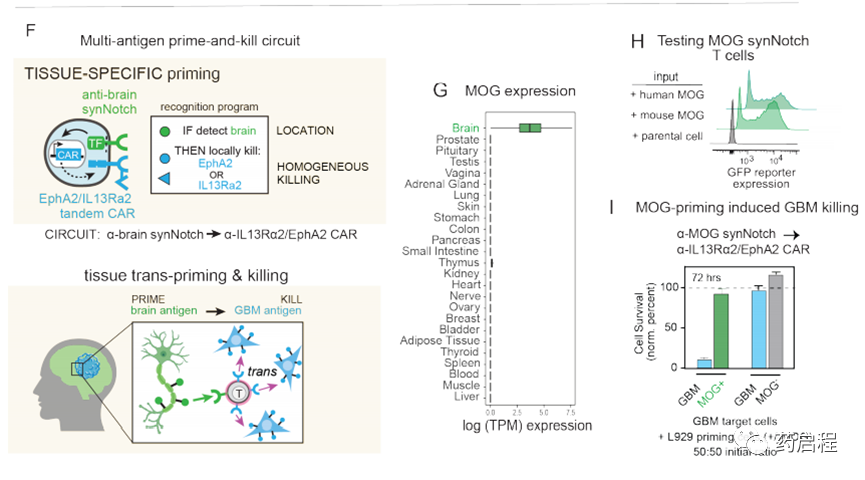
我们还假设,T细胞也可以通过识别组织特异性抗原(在非恶性细胞上表达)来局部引发(图1F)。例如,在GBM的情况下,我们可能设计由脑特异性抗原引发的T细胞回路,然后通过诱导针对GBM抗原EphA2和IL-13Rα2的CAR来触发局部杀伤。因此,脑抗原引发的回路可能提供治疗EGFRvIII阴性GBM肿瘤的解决方案。我们通过生物信息学鉴定了两种候选的大脑表面蛋白,Cadherin 10(CDH10)–一种大脑特异性的钙粘着蛋白,以及髓磷脂少突胶质细胞糖蛋白(MOG)–一种神经元髓鞘上的表面蛋白。这些抗原的预测组织表达如图1G和图S7A所示。
在本研究中,我们主要专注于使用MOG作为启动抗原,因为事实证明它具有更强的大脑特异性(图S7A中显示了CDH10作为启动抗原的分析)。我们鉴定了与MOG结合的抗体,并用它们构建了同源的synNotch受体,这些受体可以被表达MOG小鼠同工型的细胞激活(图1H),从而使这些受体可由内源性小鼠脑组织引发。然后,我们使用α-MOGsynNotchàαIL13Rα2/ EphA2 CAR回路对CD8+ T细胞进行工程改造,并在存在或不存在引发细胞的情况下将它们与GBM目标细胞(此处为GBM6 PDX细胞系)共培养(经改造的L929细胞表示MOG)。我们发现这些T细胞可以有效杀死GBM细胞,但只有在MOG +引发细胞存在的情况下(图1I)。重要的是,这些T细胞没有显示出对启动细胞的任何杀死。总而言之,这些体外研究表明,存在多种设计用于针对GBM的引发和杀死回路的策略,这些策略执行反引发/杀死,因此有可能克服抗原异质性,同时仍保持高特异性。
基于这些体外数据,我们接下来在GBM异种移植小鼠模型中评估了这些引发和杀死CAR T细胞的抗肿瘤活性。首先,我们想确认由EGFRvIII引发的T细胞在体内也可以对EGFRvIII-GBM细胞进行转杀,但仅在存在EGFRvIII+引发细胞的情况下。作为原理上的证明,我们向NCG小鼠植入了双重肿瘤–在大脑中,我们植入了U87肿瘤,其比例为50%EGFRvIII+和50%EGFRvIII-;在侧面,我们植入了U87肿瘤(仅EGFRvIII)(图2A)。在此,侧腹肿瘤代表潜在的交叉反应性正常 表达杀伤抗原但不表达启动抗原的组织;相反,脑肿瘤既具有启动抗原又具有杀死抗原。肿瘤接种后第6天,小鼠接受静脉内注射。给予初免和杀伤性CAR T细胞或对照未转导的T细胞(n=6 /组)。所有用对照T细胞治疗的小鼠均在两个部位均显示出肿瘤生长,并迅速达到安乐死终点,中位生存期为25.5天。相比之下,用原始杀伤性CAR T细胞治疗的小鼠与对照小鼠相比,表现出对颅内肿瘤生长的显着抑制(p t检验)。然而,重要的是,与对照组相比,用致敏和杀伤性CAR T细胞治疗的小鼠对侧腹肿瘤没有统计学上的显着抑制(p = 0.4;t检验,图2B)。在非引发性侧翼肿瘤中选择性缺乏杀伤作用表明,引发性和杀伤性CAR T细胞的细胞毒性活性在空间上局限于表达引发性和杀伤性抗原的肿瘤。
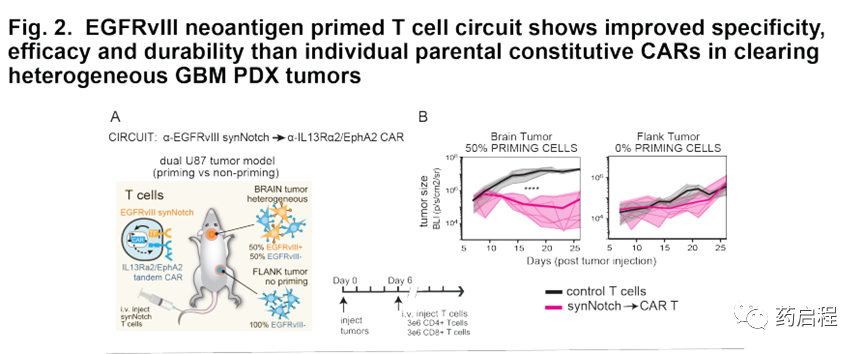
我们还对0%,50%和100%EGFRvIII+ U87细胞杀死植入的肿瘤进行了系统比较(图S3 A,B)。我们发现,致敏和杀伤性CAR T细胞未显示出0%EGFRvIII阳性肿瘤的任何清除率,但是显示了50%和100%EGFRvIII阳性肿瘤的等效清除率(对照未显示清除率)。因此,在这种情况下,引发和杀死CAR T细胞可以识别并有效克服具有异源EGFRvIII表达的肿瘤,但是可以以特异性的引发抗原门控方式进行。
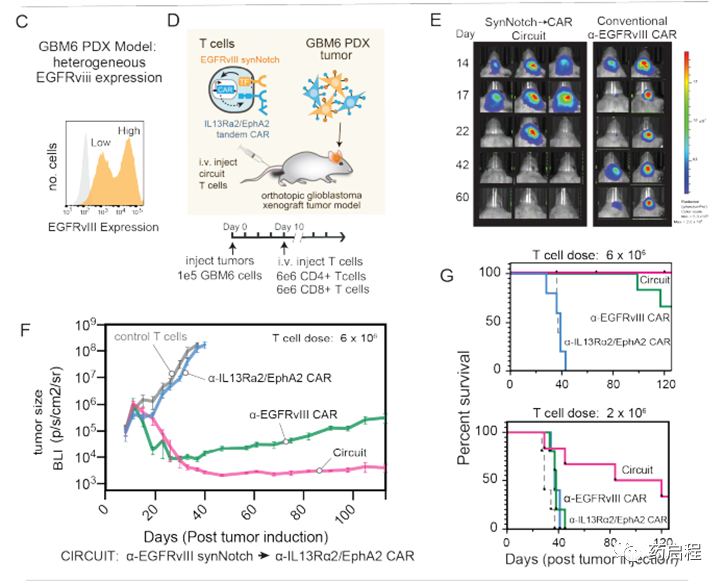
然后,我们试图评估在肿瘤模型中引发和杀死CAR T细胞的功效,该模型表现出EGFRvIII表达的天然异质性。我们将GBM6患者来源的异种移植(PDX)肿瘤鉴定为具有侵略性的GBM模型,显示出内在的EGFRvIII异质性(23)(图2C)。同样重要的是,GBM6肿瘤在体内逃避EGFRvIII单抗原CAR的治疗表现出高度可复制的能力(图2F)。当用α-EGFRvIIICAR治疗小鼠时,GBM6肿瘤急剧缩小,但随后又缓慢而稳定地复发,并具有很高的再现性(图2F,图S3C,如下所述)。这些复发性肿瘤表现出EGFRvIII表达的丧失(图2J)(我们在体外证实存在一部分GBM6细胞,这些细胞显示出无法检测到的EGFRvIII抗原,并且对常规EGFRvIII CAR的杀伤具有抵抗力-图S4 C,D)。因此,GBM6肿瘤模仿了在α-EGFRvIIICAR临床试验中观察到的基于异质性的逃逸,因此代表了一种理想的肿瘤模型,可以在其中评估可以克服这些问题的替代回路。
我们测试了带有EGFRvIII引发回路的T细胞,组成型表达α-EGFRvIIICAR或α-IL13Rα2/EphA2串联CAR的T细胞,非转导(对照)T细胞对大脑中带有GBM6肿瘤的NCG小鼠的治疗(图2F,G)。在肿瘤接种后第43天,所有接受非转导的对照T细胞的小鼠(n = 5)都死于肿瘤进展(图2E,F)。用组成型α-IL13Rα2/ EphA2串联CAR治疗在很大程度上无效(尽管这些T细胞在体外具有有效的细胞毒性)(图S4D)。用α-EGFRvIIICAR T细胞治疗可产生初始肿瘤缩小,但始终如一(n = 6)导致所有小鼠中EGFRvIII阴性肿瘤复发(6只小鼠中有3只在第125天死于肿瘤进展)(图2 E,缩略词)。与之形成鲜明对比的是,所有用原发性和杀伤性CAR T细胞治疗的小鼠(n = 6)均显示GBM6肿瘤的长期完全缓解。这种更持久,更彻底的肿瘤清除率具有很高的重现性(图S3C),这也反映在用引发和杀伤回路(多次给药)治疗的小鼠的存活率显着提高的情况下(图2G)。
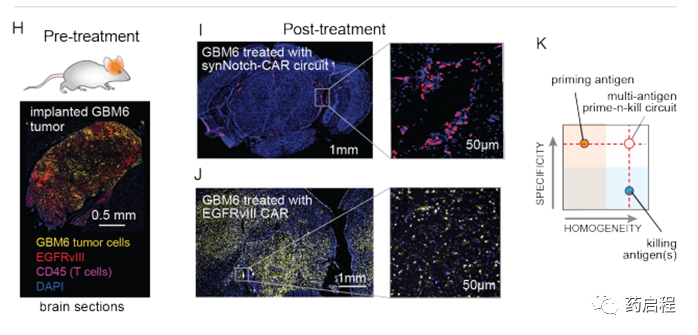
我们对用EGFRvIII CAR或引发杀伤回路处理的小鼠进行了事后免疫荧光分析。用灌注和杀伤回路治疗的小鼠的大脑显示不存在GBM6肿瘤细胞(与肿瘤清除率一致),但在脑实质和脑膜中显示出CAR T细胞(粉红色)的持久性(图2I)。相反,用EGFRvIII CAR治疗的大脑显示出大量的GBM6肿瘤细胞(黄色–与复发一致),但是EGFRvIII抗原丢失(红色),而CAR T细胞没有存活(图2J)。总之,体内肿瘤杀伤研究支持这样一个概念,即双抗原回路可以将引发抗原的特异性与杀伤抗原的同质性相结合,以实现比针对单个抗原的CAR所能实现的特异性更高但更彻底的杀伤。
为了直接观察体内T细胞启动,我们将GFP标签融合到了诱导的α-IL13Rα2/EphA2 CAR上。T细胞输注后六天,对来自受体小鼠的脑切片的分析显示,肿瘤中存在GFP+引发和杀伤性CAR T细胞(与人CD45共同染色)(图3A)。相反,在肿瘤外部(邻近脑组织)或脾脏中均未发现引发的T细胞(图3A)。此外,我们在注射后两天对这些致敏和杀死的T细胞进行了活体成像,结果显示肿瘤中有大量致敏的(绿色)T细胞,以及接近肿瘤时变为绿色的T细胞( 图3B,电影S2)。重要的是,这些电影表明,引发的T细胞稳定地定位在肿瘤内(大概与靶细胞相互作用),并且没有迅速进入或移出肿瘤。这种行为可能有助于解释这些T细胞的高度特异性和局部杀伤作用。
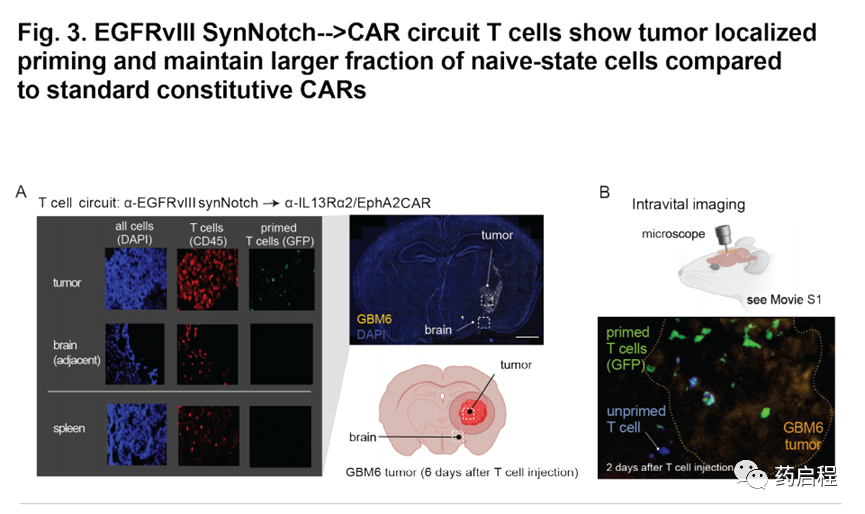
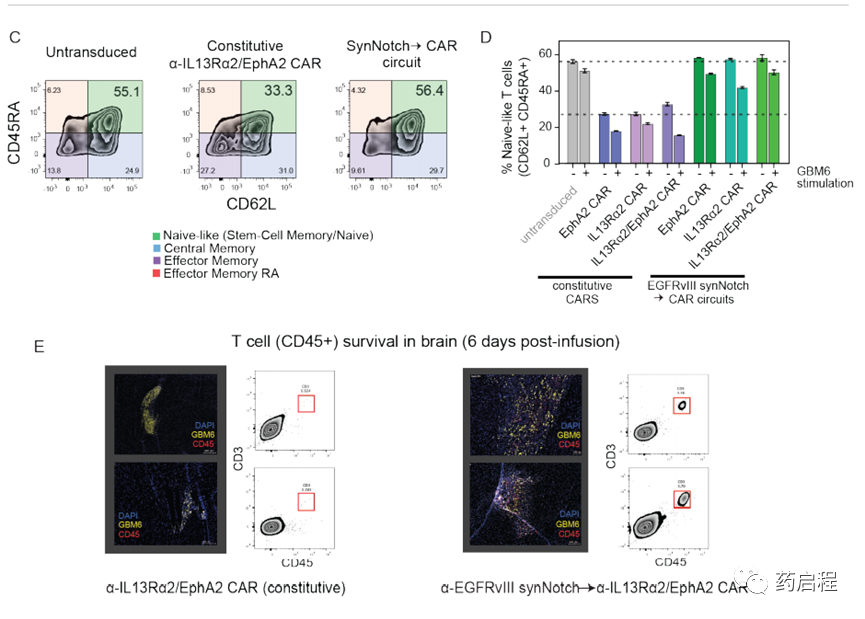
这些体内研究中最令人惊讶的发现之一是,与组成型αIL13Rα2/ EphA2 CAR T细胞相比,初免和杀伤性T细胞的肿瘤清除能力明显更好,因为两组T细胞均使用相同的CAR杀死分子,两者在体外杀死肿瘤方面同样有效。
这些观察结果表明,synNotch诱导的CAR回路具有其他功能,可显着提高体内的抗肿瘤活性。用CAR T细胞治疗实体癌的一般挑战是T细胞的耗尽,这阻止了持久的抗肿瘤活性。最近的研究表明,组成型表达的CAR的强直信号可以在提高其疲惫敏感性中起重要作用(24,25)。因此,我们通过流动分析检查了不同类型T细胞的分化状态,发现本研究中使用的所有synNotchàCART细胞在幼稚样状态(CD62L+ CD45RA+,幼稚或干中央记忆)中均显示出更高比例的细胞。)与等效的组成型CAR T细胞相比(图3C,D)。此外,当我们直接研究体内T细胞的持久性时(注入小鼠后6天),我们观察到了大量的synNotchàCAR回路T细胞。相反,我们发现此时没有存活的组成型串联CAR T细胞(图3E)。在一起,这些发现与一个简单的模型是一致的:synNotchàCAR回路阻止了通常在组成性表达的CAR中观察到的强直性信号传导,从而使T细胞保持了更幼稚的状态,不易耗尽。因此,CAR表达的局部瞬时引发不仅增加了靶向特异性,而且似乎产生了更有效和持久的T细胞状态。
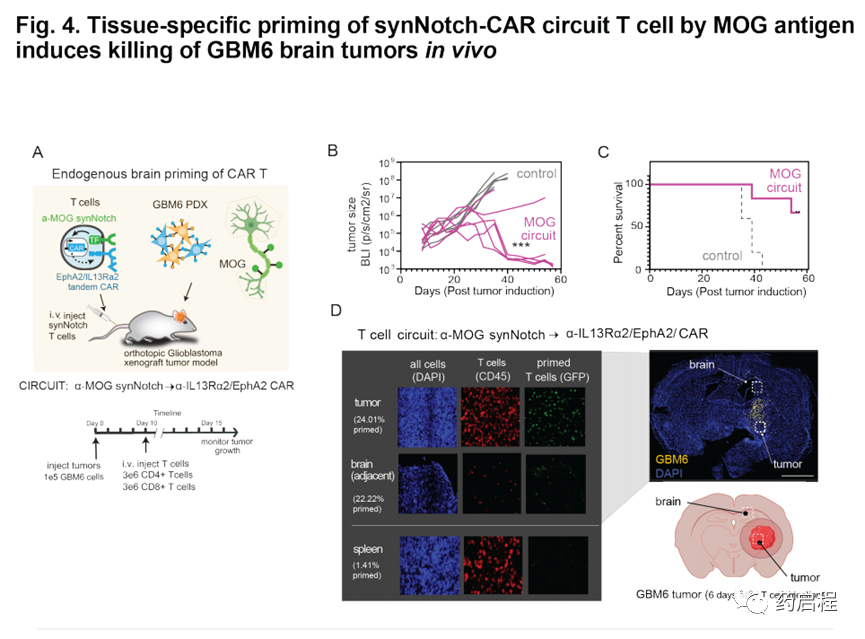
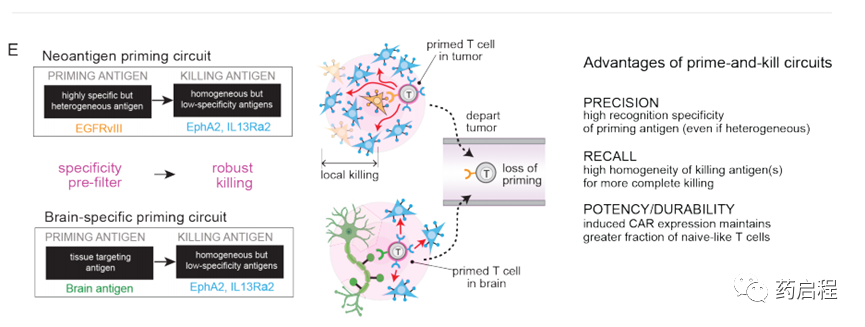
我们还想测试脑特异性抗原引发的T细胞在体内是否有效。因此,我们用植入带有α-MOGsynNotchàα-IL13Rα2/EphA2 CAR回路的T细胞治疗了植入颅内GBM6 PDX肿瘤的NCG小鼠(图4A)。GBM6肿瘤细胞不表达MOG,因此为了引发T细胞,必须通过宿主小鼠大脑内源性表达的MOG引发它们(图1I)。我们发现,MOG引发的T细胞在清除GBM6肿瘤和增加小鼠存活率方面非常有效(图4 B,C)。在将GBM6肿瘤植入侧面而不是大脑的小鼠中,MOG引发的T细胞无法清除肿瘤(图S7F),这与需要局部脑信号以许可T细胞进行杀伤相一致。用α-CDH10synNotchàα-IL13Rα2/EphA2 CAR回路治疗的小鼠也显示出对GBM6的有效杀灭,但在这种情况下,在杀死脑瘤和侧翼肿瘤方面显示出差的辨别力,这表明MOG优于CDH10,因为它严格限制了大脑的活动,特异的启动抗原。
对用MOG引发的T细胞治疗的小鼠进行的事后免疫荧光分析显示,肿瘤中存在大量的T细胞,其中许多处于引发状态(通过GFP融合CAR观察)(图4D)。在肿瘤邻近的脑组织中,我们观察到较少的T细胞,但它们也被引发(GFP+)(在脾脏中未观察到引发的T细胞)。这些结果与整个脑内的T细胞引发,推测与CAR活化和增殖性细胞因子释放导致的更明显的T细胞在肿瘤中的扩散相一致。
这种脑特异性抗原引发回路的有效性代表了工程治疗细胞的新进展-该回路不仅代表了治疗EGFRvIII-GBM的可能方法,而且还可能为工程针对脑的细胞疗法提供一般策略 治疗广泛的神经系统疾病,包括其他脑部肿瘤,神经炎症或神经退行性疾病。
讨论
总之,这些结果表明,有多种方法可以设计synNotchàCAR回路,这些回路可以有效地结合识别不完美的互补抗原作为单个抗原靶标。可以建立基于高度特异性的新抗原(如EGFRvIII)或组织特异性抗原(如MOG)引发的回路。至关重要的是,这些引发抗原不需要全部存在于所有肿瘤细胞上,也不必全部存在于任何肿瘤细胞上(在组织特异性引发的情况下)。一旦启动,就可以对T细胞进行编程,使其靶向肿瘤上同质的CAR抗原,从而完成完整的肿瘤杀死,即使它们自身具有不完善的特异性。通过整合来自多种抗原和多种细胞的信息,这些回路从本质上为我们提供了将肿瘤细化为复杂组织的改进能力,从而为如何以更安全,更具体的方式识别和攻击肿瘤开辟了许多新的可能性。其他相关策略则将CAR和双特异性衔接子结合起来以整合多抗原组合(26)。
我们在这里显示synNotchàCART细胞具有将其与常规CAR T细胞区分开的多个特征,这可能证明在治疗实体癌(例如GBM)方面具有很高的优势。首先,它们的多抗原识别增强了肿瘤细胞与正常细胞之间的区别。其次,这些回路介导反式启动/杀死的能力使它们能够克服靶抗原异质性的逃逸。最后,将CAR表达置于受控控制之下的简单行为似乎将T细胞维持在更加幼稚的状态,这种状态更加持久且不易疲劳。对于针对除GBM以外的其他癌症的synNotchàCAR T细胞,观察到了类似的改善的抗肿瘤活性(27),这表明这些回路可能为治疗许多实体癌提供了非常有效的一般策略。


D
O
i
:
Multi-antigen recognition circuits overcome challenges of specificity, heterogeneity, and durability in T cell therapy for glioblastoma
https://doi.org/10.1101/2021.01.07.425632
编辑:小果果,转载请注明出处:https://www.cells88.com/cells/gxb/9189.html
免责声明:本站所转载文章来源于其他平台,主要目的在于分享行业相关知识,传递当前最新资讯。图片、文章版权均属于原作者所有,如有侵权,请及时告知,我们会在24小时内删除相关信息。
说明:本站所发布的案例均摘录于文献,仅用于科普干细胞与再生医学相关知识,不作为医疗建议。
 微信扫一扫
微信扫一扫  支付宝扫一扫
支付宝扫一扫 